Hypophosphatasia plod. Orogenic displazija
hypophosphatasia To je redka avtosomno recesivna dedna patologija, ki je značilna demineralizacije kosti in nizko alkalne fosfataze v serumu in drugih tkiv. Delovanje alkalne fosfataze v pirofosfata in druge kompleksne fosfoefiry vodi do kopičenja anorganskega fosfata, ki so pomembni za tvorbo kosti trabekul. Krhkost kosti v tej bolezni, je le posledica pomanjkanja razvoja.
hypophosphatasia Razdeljen je na tri kliničnih vrst, odvisno od starosti, ko se začne bolezen: prirojeno, novorojenčkov, otrok in odraslih hypophosphatasia. Prirojeno, neonatalno in v večini primerov otroške oblike imajo avtosomno recesivno dedovanje, medtem ko oblika za odrasle - avtosomno dominantnantny.
V prirojene neonatalne-varianti te bolezni pogosto pojavi smrt zarodka ali smrtjo novorojenčka.
Sadje z prirojena hypophosphatasia imajo posplošeno demineralizacije skeleta s skrajšanjem in ukrivljenost dolgih kosti. Obstaja več zlomov. Pomenilo demineralizacijo lobanjsko oboka, kar ima za posledico deformacijo obliki glave, če imate zunanji pritisk na njej. Echographic To funkcijo je mogoče zaznati tudi v nekaterih primerih, osteogenesis imperfecta tipa II in tipa ahondrogeneza 1A.
V literaturi obstajajo znaki pred rojstvom diagnostika Ta bolezen z ultrazvokom in analizirali raven alkalne fosfataze v tkivni kulturi horionskega resic in amnijske celice tekočin. Merjenje alkalne fosfataze v amnijske sebi tekočine ni zanesljive metode diagnoze hypophosphatasia, saj je glavni del tega encima v plodovnici z delovanjem plodu gastrointestinalnega trakta povzročena.

Encimi, ki se pojavi nenormalnosti pri hypophosphatasia, zastopnika alkalne fosfataze jetra in kosti ter povzročila le 16% celotne encimsko aktivnost amnijske tekočine.
Zdaj je mogoče izvesti pred- diagnostika otroci tip hypophosphatasia uporabo molekularnih genetskih študij horionski villus tkiva. Obstaja opis primera, ko je bila ugotovljena missense mutacije pri pregledu novorojenčka s tem pogojem.
Orogenic displazija je avtosomno recesivna bolezen, označen s micromelia, clubfoot, razvojne motnje ščetke, več upogibnim kontrakture sklepov in skoliozo. Zaradi variabilnosti fenotipskih manifestacij svojih diagnostike ob rojstvu je lahko težavno, in v primerih zmerne resnosti diagnozo patologije je določena kasneje.
klinične manifestacije rizomelicheskogotipa vključujejo skrajšanje udov, kontraktur, krtače napake z namestitvijo palca v položaju ugrabitve (palec "štoparja» - štoparja palca) in hudo nenormalno namestitev stop tipa "konja peš".
struktura glava ponavadi niso spremenile, vendar pa lahko mikrognatija in volčje žrelo. Ta vrsta displazije spremlja generalizirane patologije hrustančnega tkiva z okvaro stanju njene medcelične snovi in tvorbe vezivnega brazgotin tkiva in njihovo nadaljnjo kostenitev. To je ta postopek omogoča nastanek kontraktur. Na bolezen posledica mutacij DTDST gena, zaradi česar je moten fosforilacijskih proteoglikanov.
 Prirojena idiopatski hiperkalciemija in hypophosphatasia. krhke kosti
Prirojena idiopatski hiperkalciemija in hypophosphatasia. krhke kosti Znaki osteogenesis imperfecta. Osteopetroze bolezen marmor kosti, bolezni Albers - Schönberg
Znaki osteogenesis imperfecta. Osteopetroze bolezen marmor kosti, bolezni Albers - Schönberg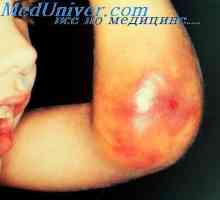 Afibrinotenemiya angiohemophilia in novorojenčkov. Hemoragična bolezen novorojenčka
Afibrinotenemiya angiohemophilia in novorojenčkov. Hemoragična bolezen novorojenčka Razvrstitev ahondrogeneza plod. diagnoza ahondrogeneza
Razvrstitev ahondrogeneza plod. diagnoza ahondrogeneza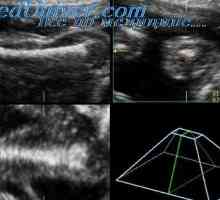 Kampomelicheskaya displazija plod. Diagnoza kampomelicheskoy displazija
Kampomelicheskaya displazija plod. Diagnoza kampomelicheskoy displazija Osteogenesis imperfecta plod. Vzroki in diagnoza osteogenesis imperfecta
Osteogenesis imperfecta plod. Vzroki in diagnoza osteogenesis imperfecta Fibrohondrogenez in atelosteogenez. Ahondrogenez ali anosteogenez
Fibrohondrogenez in atelosteogenez. Ahondrogenez ali anosteogenez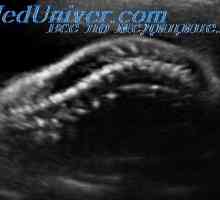 Hipoplaziji od prsnih celic zarodka. Hondroektodermalnaya displazija
Hipoplaziji od prsnih celic zarodka. Hondroektodermalnaya displazija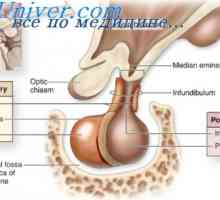 Prirojena skrajšanje stegnenico. Cepitev na rokah in nogah za plod
Prirojena skrajšanje stegnenico. Cepitev na rokah in nogah za plod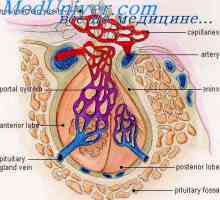 Pomanjkljivosti zmanjšanje udov pri plodu. Fokomelija plod
Pomanjkljivosti zmanjšanje udov pri plodu. Fokomelija plod Akromezomelicheskaya displazija. eykardi sindrom
Akromezomelicheskaya displazija. eykardi sindrom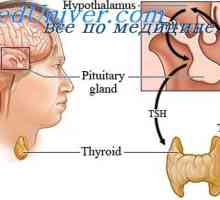 Hypophosphatasia. Hipoplastična levo srce
Hypophosphatasia. Hipoplastična levo srce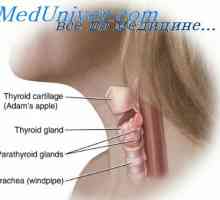 Mezomelicheskaya displazija. Diagnoza in prognoza mezomelicheskoy displazija
Mezomelicheskaya displazija. Diagnoza in prognoza mezomelicheskoy displazija Mikrotsefalichesky primarni pritlikavost. Sindrom neurejeno-Lax
Mikrotsefalichesky primarni pritlikavost. Sindrom neurejeno-Lax Osteogenesis imperfecta. Diagnoza in Prognoza osteogenesis imperfecta pri plodu
Osteogenesis imperfecta. Diagnoza in Prognoza osteogenesis imperfecta pri plodu Tanatofornaya displazija. Diagnoza in prognoza tanatoformnoy displazija
Tanatofornaya displazija. Diagnoza in prognoza tanatoformnoy displazija Avtosomno recesivno dedovanje. -X vezano dedovanje
Avtosomno recesivno dedovanje. -X vezano dedovanje Avtosomno recesivna policističnih ledvic pri otrocih. Diagnoza in zdravljenje
Avtosomno recesivna policističnih ledvic pri otrocih. Diagnoza in zdravljenje Podlakti sev
Podlakti sev- Clubfoot deformacija stopala, da se obrnejo navznoter in proti podplatih. Vzroki clubfoot: kosti…
 Drugo patologija pri novorojenčkih
Drugo patologija pri novorojenčkih