Zlatenica otrok na pomanjkanje alfa-1-antitripsina (a1-am)
Alfa-1 antitripsin (A1-AT) je velik zaviralec plazme proteaze, ki nadzoruje proteolitično aktivnost nevtrofilne elastaze, hidrolizo strukturne beljakovine. Ti Pi serin protein, znan kot serpinov, so vključeni v regulacijo proteolize med koagulacijo, fibrinolizo in vnetja z inhibicijo aktivnosti proteaze in destruktivne nevtrofilne elastaze.
Gene alfa-1-antitripsin (A1-AT) - je gen sestoji iz nukleotidnih zaporedij 12 kilobazami velikosti, ki se preslikajo na 14. kromosomu. Znano je, da ima veliko možnosti, vključno normalen alel M. Približno 2% novorojenčkov sta A1-AT s primanjkuje genaa1 Z-alel-AT. Ta varianta je povezano z zmanjšanjem ravni A1-AT v serumu.
nizka stopnja alfa-1-antitripsin (A1-AT), in je lahko klinično pomembne pri drugih izvedbah, kot S-alela in tipa "nič", ki pa zgodnji pojav jetrnih bolezni le značilnost homozigotna pi ZZ. Homozigotnosti Pi ZZ je relativno pogosta patologija. Njegova frekvenca je 1 primer na 2000 živorojenih otrok. Opredeliti različne stopnje spremembah a1-AT uporablja metodo delcev po.
Na podlagi te analize, klasifikacije vrst migracij v električnem polju. Običajno varianta M migrira v srednji M, medtem ko nenormalne variante A, L migrirajo naprej in variante N, Z migrirajo počasneje. Z-alel A1-AT gena razlikuje od običajne M-alel zamenjavo ene same gvanin do adenin na kodonu GAG glutamina, da spremembe ostankov lizina.
Nenormalno vsebujejo Z-alel alfa gen 1-antitripsin (A1-AT) drži v endoplazemski retikulum hepatocitov in se kopiči v celici, in posledično se zmanjša na 80-85% stopnjo alfa-1-antitripsina (A1-AT) v serumu. Najbolj pridržan v celici A1-AT razpade, temveč tvori preostalo količino znotrajcelično kopičenje netopnih proteinov iz vključitve A1-AT preoblikovati. Ti grozdi ne more slediti poti izločanja.
Kljub pomanjkanju jasne razlage za ta pojav, je znano, da je vključitev podatkov povzroči škodo gepatotsitov- pride le 17% dojenčkov s homozigotno ZZ a1-AT klinično pomembno jetrno bolezen v otroštvu. Vendar pa je pomanjkanje A1-AT je najpogostejši genetski vzrok bolezni jeter pri otrocih in najpogostejših genetskih bolezni, o katerih izvajajo transplantaciji jeter pri otrocih.

Obstajajo špekulacije, da je osnova za kopičenje mutanta alfa-1-antitripsin (A1-AT) v retikulumu kar je povzročilo jeter patologija lahko leži katerokoli genetske vzroke. Leta so študije mobilnega transdukcija zaznali znatno zamudo razkroj mutant A1-AT po kopičenje fibroblastov v skupini bolnikov homozigoti ga je ZZ, imeli bolezen jeter, v primerjavi s homozigotno bolnikih brez bolezni jeter.
Glede na rezultate nedavne študije so predlagali morebitno vpliv proteazoma aktivnosti v citoplazmi omenjenega procesa razpada. Ni dokazov, da so imunoregulacijsko geni vpleteni v patogenezo poškodbe jeter, povezane z a1-AT.
Trenutno presaditev jeter je edina možna oblika zdravljenja otrok s hudo boleznijo jeter s pomanjkanjem alfa-1-antitripsina (A1-AT) povzročajo. Starejša starost bolnikov s pomanjkanjem A1-AT v prisotnosti emfizema (stanja, ki se ne pojavlja pri otrocih) bolj konzervativnim (netransplantatsionnaya) terapija obsega dajanje a1-AT intravensko ali aerosol, uporaba inhibitorji nevtrofilne elastaze in prenos genov preko virusne prevozniki.
Nadomestno zdravljenje z alfa-1-antitripsin (A1-AT) v obliki intravenske pripravku problematična zaradi visokega tveganja in morebitnih zapletov pri uporabi izdelkov človeške plazme. Raziskave te vrste zdravljenja so izvedli le pri odraslih bolnikih. Trenutno obstajajo načini, kako hitro pridobitev rekombinantnih oblik a1-AT iz človeške krvi, zaradi česar pripravki A1-AT so na voljo zdaj. Vodenje raziskali možnost uporabe genskih tehnik za zmanjšanje ali odpravo primanjkljaja a1-AT.
Običajna gen alfa-1-antitripsin (A1-AT) oseba je bil uspešno izveden v progastih mišičnih celicah uporabo adenovirusa-nosilec pri poskusih na živalih. Študije pri človeški populaciji naprej.
Natančno mogoče predvideti resnosti poškodbe jeter pri dojenčkih z pomanjkanje alfa-1-antitripsina (A1-AT) je odsoten. Pogosto kot prvi simptom jetrne patologije pri otrocih v prvih mesecih življenja označiti dolgo zlatenico. Majhno število otrok razkrije hepatosplenomegalija z ascitesa in motnjami v delovanju jeter. Nekateri bolniki s pomanjkanjem a1-AT v neonatalnem obdobju razvije fulminantni odpovedi jeter. Starši bolnih otrok, je treba opraviti genetsko svetovanje v zvezi z možnim tveganjem za Pi ZZ v naslednjih nosečnostih.
Ocenjuje se, da bo približno 72% sorojencev teh probands z Pi ZZ jetrne bolezni, 29% jih bo hude poškodbe jeter. Zato je zaželeno, da se Porodničarstvo horionbiopsiyu namen prenatalne diagnoze pomanjkanja a1-AT. Za odkrivanje sprememb v nukleotidni sekvenci mutacij Z kraja na pomanjkljivost a1-AT sprejemljivo metodo PCR.

 Analize v kronični pankreatitis kri, blato
Analize v kronični pankreatitis kri, blato Prevladujoči in recesivni alelov kromosomov. Avtosomalne dominante dedovanje
Prevladujoči in recesivni alelov kromosomov. Avtosomalne dominante dedovanje Drugi beta-adrenergični blokatorji pri zagotavljanju prve pomoči
Drugi beta-adrenergični blokatorji pri zagotavljanju prve pomoči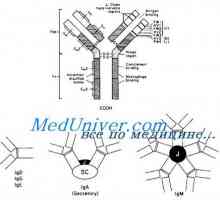 Zapletanje težke verige protitelesa geni. Geni težke verige IgA
Zapletanje težke verige protitelesa geni. Geni težke verige IgA Pomanjkanje mieloperoksidaze (MPO). Klinika in diagnostika
Pomanjkanje mieloperoksidaze (MPO). Klinika in diagnostika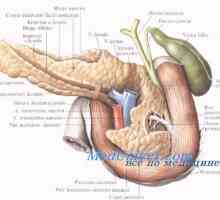 Metode vrednotenja eksokrini pankreas
Metode vrednotenja eksokrini pankreas- Nova tarča za zdravljenje raka na dojki
- Anatomija, prekrvavitev in oživčenje trebušne slinavke
- Alfa-1-antitripsina pomanjkljivost, alfa (a) z nizko molekulsko maso -antitrypsin inhibitorja…
- Meflokin (meflokin) dl-eritro-alfa, 2-piperidil-2,8-bis (trifluorometil) -4-quinolinemethanol.…
- Permikson (perimxon) ekstrakt (lipidosterinovy), ki jih tolažila ventilatorja-dlani (Serena…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Hematologija-vpliv epoetin alfa o parametrih in kakovost življenja pri bolnikih z rakom, ki…
- Terapija-defetsit inhibitor proteaze dejavnik tveganja alfa-1-antitripsina pri razvoju in…
- Terapija-pankreasa bolezen
- Črevesne bakterije zaščito pred malarijo
- Celični protein TLR5 - ključ za zdravljenje revmatoidnega artritisa
 Alfa pomanjkljivost 1 antitrypsin
Alfa pomanjkljivost 1 antitrypsin 1-Antitripsin pomanjkljivost, -fetoprotein, -fetoprotein in jetrne bolezni,-verige bolezni
1-Antitripsin pomanjkljivost, -fetoprotein, -fetoprotein in jetrne bolezni,-verige bolezni Klinična Elektroencefalografija (EEG)
Klinična Elektroencefalografija (EEG)