Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija
DAX1
--X vezan gen, ki je izražen v razvoju zarodek. Posebni organi njegov izraz - hipotalamusa, hipofize, nadledvične žleze in spolne žleze. Kršitve DAX1 izraz kaže na razlike med tlemi in prirojene nadledvične hipoplazije. DAX1 - beljakovina jedrska receptor, dobila ime iz angleške kratice:• D (dozirna občutljiva spola preobrat) - od odmerka odvisno neusklajenost tla;
• s (nadledvične hipoplazija prirojena) - prirojena adrenalna hipoplazija;
• X - kritično območje kromosoma X;
• 1 - 1 gen.
očitno, DAX1 Spada v skupino genov antagoniste moškega tipa. Mutacije ali delecije DAX1 gen vzrok prirojena adrenalna hipoplazija. Podvajanja tega gena vodi k razvoju neenakosti spolov pri fenotipskih ženskah z XY genotip. DAX1 genska mutacija odgovoren za nastanek prirojenih nadledvične hipoplazije z nadledvične razvoj hipofize nedostatochnostyu- patologijo vodi hipogonadotropnega hipogonadizma.

razlika med spoloma XY genotip
podvajanje regija DAX1 lahko privede do neusklajenosti spola in razvoja genotip XY blok testisov (gonadno dysgenesis). Gonadni, ki ne vsebuje spermo ali jajčne celice, ki se imenuje poloskovidnoy Gonadni. Delno gonadne dysgenesis lahko razvije tem pa ohraniti funkcionalno aktiven del testisov tkiva. Kjer lahko zunanjih genitalij je biseksualec. Fenotipa moški z razlikami spola označen z odsotnostjo Mullerian zaviralni dejavnik in vztrajnosti Müllerian. V tem se razlikujejo od feno-tipično ženske z XY genotipom, androgene neobčutljivosti, ki nimajo preostale Mullerian struktur.
Bolniki s čisto gonadno dysgenesis XY genotip večjo gonadoblastomy tveganja, ki jih je naletel na frekvenci 10-30%. Večina primerov čistega žlez dysgenesis s mutacij SRY gen povzroča. Raziskava teh bolnikov mora vključevati analizo kariotip za kromosomske izbrisov X, sry gensko analizo za mutacije in FISH-molekularne ali testiranja za genske podvajanja DAX1. V zapletenem zdravljenje mora vključevati estrogensko nadomestno zdravljenje zapoznelo puberteto.
Kampomelicheskaya displazija in SOX9 genske mutacije
SOX9 gen je bil opisan je leta 1994 kot regulator spolne diferenciacije moškega tipa, ki sodelujejo pri razvoju skeleta procesih. SOX9 gen se nahaja na daljšem kraku kromosoma 17 in do neke mere homologna gena SRY, od tod tudi njeno ime - SOX, tj "Polje, povezane s tlemi."
Deluje kot faktor transkripcije in je izražena v človeku na modih, ledvic, hondrocitov, jeter in možganov. SOX9 igra ključno vlogo v odvisnosti od odmerka spolne diferenciacije moškega tipa, ki je bil prikazan v haploinsufficiency SOX9, ki vodi bodisi neskladje spola, bodisi ambivalentne spolne diferenciacije. Povečevanje odmerkov SOX9 zaradi podvajanja povzroča avtosomnem razlik med spoloma pri osebah z genotipom XX. Poleg tega je SOX9 vključen v aktivacijo Mullerian zaviralni dejavnik, kot tudi igra pomembno vlogo pri razvoju kosti in hrustanca, verjetno kot regulator ekspresijo COL2A1, kot je prikazano pri bolnikih z nepravilnostmi skeletne displazije pri kampomelicheskoy.
Kampomelicheskaya displazija - sporadična motnjo značilno kratke postave, kratkih krakov, upognjenih spodnjih okončin, oslabljeno strukturo obraza, razcepljenim nebom in pogosto prirojeno srčno boleznijo. Pogoste smrti, povezanih z mutacije SOX9 gena. Menijo, da se bolezen deduje avtosomno dominantnega, z visoko frekvenco za to de novo mutacije zaradi visoke smrtnosti. Kot veliko drugih sindromov, pri katerih obstaja sum spolnih žlez Mozaicizem, tveganje ponovnega pojava bolezni za par je približno 3%.
 Nepravilnosti SOx genov in TVH Holt-Oram sindroma. Dejavniki fibroblastov rasti
Nepravilnosti SOx genov in TVH Holt-Oram sindroma. Dejavniki fibroblastov rasti Mezomelicheskaya displazija. Diagnoza in prognoza mezomelicheskoy displazija
Mezomelicheskaya displazija. Diagnoza in prognoza mezomelicheskoy displazija Prirojena nadledvične hiperplazija. Zdravljenje nadledvične hiperplazijo.
Prirojena nadledvične hiperplazija. Zdravljenje nadledvične hiperplazijo. Oskrbovanje v nadledvične žleze neuspeh: epidemiologija bolezni
Oskrbovanje v nadledvične žleze neuspeh: epidemiologija bolezni Hipofiza pritlikavost ali infantilizem. Pritlikavost ali acromicria
Hipofiza pritlikavost ali infantilizem. Pritlikavost ali acromicria Addisonova bolezen. Kronična insuficienca nadledvične žleze
Addisonova bolezen. Kronična insuficienca nadledvične žleze Virilizing prirojene nadledvične hiperplazija: Vzroki in mehanizmi razvoja
Virilizing prirojene nadledvične hiperplazija: Vzroki in mehanizmi razvoja Diferencialna diagnoza in zdravljenje virilizing nadledvične hiperplazijo
Diferencialna diagnoza in zdravljenje virilizing nadledvične hiperplazijo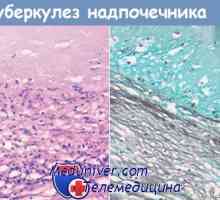 Morfologija kronične insuficience nadledvične žleze. Patološka anatomija nadledvične tuberkuloze
Morfologija kronične insuficience nadledvične žleze. Patološka anatomija nadledvične tuberkuloze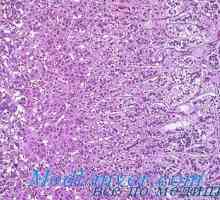 Morfologija nadledvičnih sifilis. Patološka anatomija nadledvične hipoplazije in atrofijo
Morfologija nadledvičnih sifilis. Patološka anatomija nadledvične hipoplazije in atrofijo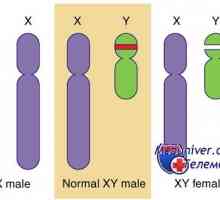 Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha
Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha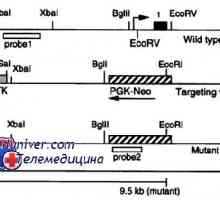 Mutacije gonadotropin receptorje. Nepravilnosti LH in FSH receptorjev
Mutacije gonadotropin receptorje. Nepravilnosti LH in FSH receptorjev Insuficienca nadledvične žleze: vzroki, klinična in diagnostika
Insuficienca nadledvične žleze: vzroki, klinična in diagnostika Bolezni endokrinega sistema in stres
Bolezni endokrinega sistema in stres Nadledvične žleze
Nadledvične žleze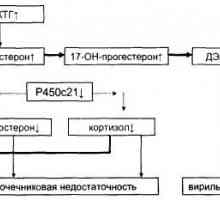 Posebej prirojena adrenalna hiperplazija
Posebej prirojena adrenalna hiperplazija Nadledvične virilization
Nadledvične virilization Določitev obloge (vohun kot določitev faktorja testisov)
Določitev obloge (vohun kot določitev faktorja testisov) Virilism, sindrom moškost
Virilism, sindrom moškost Prirojena nadledvične hiperplazija: simptomi, zdravljenje, diagnoza
Prirojena nadledvične hiperplazija: simptomi, zdravljenje, diagnoza Sekundarni insuficience nadledvične žleze: Simptomi, zdravljenje, diagnoza, vzroki
Sekundarni insuficience nadledvične žleze: Simptomi, zdravljenje, diagnoza, vzroki