Genetski sindromi, assotsiirovannn z feokromocitom in paraganglioma

Vsebina
Večina feokromocitom - občasne, čeprav se ugotovi, da je veliko število teh tumorjev somatskih mutacij podobne zarodka (povezane sorodstvene sindrom).
Na podlagi podatkov iz družinske zgodovine, preden so verjeli, da se le 10% bolnikov s takšnimi tumorji del genetskih sindromov. Vendar pa se z uvedbo metod genetska analiza je pokazala, da je 20-30% bolnikov z feokromocitom in paraganglioma nosilci zarodne mutacije, povzroča razvoj družinskih sindromov. Tako je približno 9% bolnikov z enostransko feokromocitom, pri kateri tumor kaže občasne mutacije zaznane VHL (von Hippel-Lindau gen bolezen). Zato je genetska analiza priporočljivo pri vseh bolnikih s feokromocitom in paraganglioma, še posebej, če extraadrenal paragangliomas, tumorji multifokalne, če se simptomi v mladosti (50 let) ali v prisotnosti tumorja v družinsko zgodovino. Genetske študije, opravljene pri bolnikih z drugimi podobam dednih sindromov, ki poskušajo odkriti mutacije proto-onkogensko RET (II MEN sindrom) gen VHL (von Hippel-Lindau bolezen), NF-1 (nevrofibromatoza) in LDH genov (SDGV in SDGD SDGS).
Multiple endokrine neoplazije tipa 2 (Men II)
Osnova avtosomnem prevladujočega sindrom Man II (rak medularni ščitnice, feokromocitom, in številnih drugih bolezenskih manifestacij) leži aktiviranju pro-toonkogena RET mutacije se nahaja na kromosomu 10, ki kodira transmembranski receptor tirozin kinaze, ki je izražen v tkivu, ki izvirajo iz nevralnega grebena. Obstajata dva podtipa tega sindroma - Men Pa (90%) in moških lib (10%). Pri bolnikih z sindromom MEN Pa označen z množico missense mutacije vodijo do zamenjave ene aminokisline v zunajcelični domeni receptorja tirozin kinaze molekul, ki so odgovorne za njegovo homodimerizacijo in aktivacijo. Pri bolnikih z moškimi sindromom llb Zaznali missense mutacijo (kodon 918 eksona 16), ki spreminja strukturo intracelularno tirozin kinaze katalitične domene in tudi vodi v njeno aktivacijo. V obeh podtipov sindrom se navadno pojavi feokromocitoma v nadpochechnikah- extraadrenal paraganglioma so redki. Hipertenzija je običajno označen s krčevitih več. Vsaka mutacija določa vrsto fenotipa (starost ob nastopu in agresivnosti medularni rak ščitnice).
Konstitutivna aktivacija tirozin kinaz mutacij na povzročijo le nekaj eksoni RET. Ponavadi vpliva eksonov 10, 11, 13, 14 in 15. Zato se lahko genetske študije omejen le na te eksonov. Če se mutacije ne zazna, potem pa v posebnih laboratorijih zaporedje preostalih 15 eksonov. Ko je mutacija v družini znana vnaprej, je genetska analiza določenega bolnika iz družine omejene na iskanju te mutacije.
Večina primerov je sindrom feokromocitoma MEN II don zaradi mutacij genov 634 RET. Bolniki s temi mutacijami je potrebno upoštevati in preučiti zelo previdno, da bi opredelili feokromocitoma. Ker v teh primerih, je ta tumor razvije v zgodnjem otroštvu (približno 5 let), je treba začeti z iskanjem za otroka redno meriti krvni tlak, in določanju višine Metanephrine plazme.
Feokromocitom so značilne za vse družine z MEN sindromom II, razen povezana z mutacijami RET gena na kodonoma 609, 768 in 891. val804met Bolniki s temi mutacijami lahko pregledajo manj pogosto.
Metastaze s fenokromocitoma MEN-sindrom II na voljo le v približno 4% primerov, kar je verjetno povezano z zgodnjim odkrivanjem teh tumorjev. Dvostranski feokromocitom najdemo skoraj 70% bolnikov, vendar je prej začetek raziskavo, bolj pogosto najdete enostransko tumor. Ko se pojavi enostransko adrenalektomiji nadledvične feokromocitoma na sekundo, približno 50% bolnikov (povprečna starost 12). Adrenalne feokromocitoma pri MEN-sindrom II izločajo noradrenalina in adrenalina (in njegov metabolit metanephrine). Vsebnost kateholaminov v plazmi lahko normalno, vendar raven Metanephrine navadno poveča. Zato, da je slika je najboljši pokazatelj majhnih feokromocitom. V nekaterih primerih pa je mogoče zaznati feokromocitoma le, če je ločeno določanje metanephrines, kateholaminov in kreatinina v vsakdanjem urinu.
- MEN IIa (Sipple sindrom). V tem sindromu se medularni rak ščitnice odkrili pri 95-100% bolnikov, hiperparatiroidizmom (zaradi hiperplazija obščitnicah) - 35%, in feokromocitom ali hiperplazije nadledvične sredici - 50% (različne od 6% do 100% v različnih družin ). Ti bolniki so zelo pogosto najdemo kot lihen planus in Hirschsprung bolezni. Feokromocitoma ponavadi pojavijo v srednjih letih, in ne vedno spremlja arterijske hipertenzije. RET proto-onkogena analiza v družinah z MEN sindromom Pa bi morala biti do 6 let. To vam omogoča, da ugotovi, da je treba za profilaktično tiroidektomije in skrbno raziskave, namenjene ugotavljanju feokromocitoma in hiperparatiroidizma.
- MEN IIb. Ta sindrom običajno vključuje medularni rak ščitnice, več neuromas sluznice (vidna za oči in usta), zadebelitev roženice živcev, ganglionitis-ROM črevesne marfanoidnuyu videz in feokromocitoma ali hiperplazije nadledvične sredice. Medularni rak ščitnice pojavi bolj agresiven in razvija pri nižji starosti kot pri moških sindrom Pa. Možnost družina mutacij prevoznikov RET proto-onkogena pri pacientu, je treba takoj preveriti po odkritju feokromocitom. Pri ugotavljanju takšnega mutacijo je profilaktično tiroidektomija. Vse medijske ustrezne mutacije zahtevajo skrbno zdravniški nadzor in pregled.
Von Hippel Lindau bolezen (BGL)
BGL - podedoval kot avtosomno dominantnega lastnost, da nagnjenost k razvoju kožnega raka prevoznike mnogih tkivih. Vendar feokromocitoma je značilna le za bolnike z diabetesom LGL 2. V nasprotju z občasnimi feokromocitom, v teh primerih, je manj maligni, redko lokalizirano izven nadledvične žleze, najpogosteje se pojavi na obeh straneh in pri nižji starosti. Povprečna starost njenih manifestacij pri bolnikih s tipom LGL 2 - 28 let, in najnižjimi - 5 let.
BGL pojavi s frekvenco 2-3 primerov na 100.000 prebivalcev. Bolniki pogosto pojavijo multicentrične hemangioblastoma (angioma) v mrežnici, malih možganov in hrbtnega cist rebrasti Očitno celice ledvičnega raka, več cist in nevroendokrinih tumorjev nedelovanja trebušne slinavke. Endolymphatic sac tumorji privede do izgubo sluha, omotico in ataksijo. Veliko žensk z BGL v jajčniku, širok ligament iz maternice, vagine, materničnega vratu in sramnih ustnic našel cystadenoma verjetno mezonefralnogo izvor. Pri moških, ki so enako cystadenoma od epididimisa.
Za genetske analize otrokovih prvih tednih življenja, je kri (v epruveto z EDTA), pošlje v laboratorij, kjer se DNK limfocitov, da razišče VHL genske mutacije. Otrok, rojen v družini s BGL, ki iščejo že znano mutacijo. Če sumite, da je otrok BGL iz družine z neznano mutacijo izvaja neposredno zaporedje celotne regije, ki kodira za VHL gena in spojnimi mestih pri iskanju točkovnih mutacij.
Klinična diagnoza LGL je določena v tistih primerih, ko bolnik iz družine z znanim mutacije tega gena pojavi eden od njihovih tumorjev, značilne bolezni. V odsotnosti družinsko anamnezo BGL njegovo domnevno diagnosticirana, ko zazna, ali iz dveh ali več hemangioblastomas, eno od njih v povezavi s feokromocitom ali raka jasno ledvic. BGL je treba sum tudi pri bolnikih z več tumorjev, ki se kaže v razmeroma mladih letih (manj kot 50 let za feokromocitom ali hemangioblastoma in manj kot 30 let, za jasno celic raka ledvic). V takih primerih, VHL gen zaporedja.
Gene VHL (zaviralnih) lokalizirana na kromosomu 3 (Zr26-25 segment) kodira dve različni proteini sestojijo iz 213 in 160 aminokislinskih ostankov. Oba sta vključeni v dejavnikih uničevanje hipoksija (HIF-1 in HIF-2) je inducirala - domene temi proteini vežejo na elongin in domene - hidroksilirane HIF (Reakcijsko zahteva prisotnost kisika). Ta kompleks nato pripisuje ubikvitin, ki omogoča znotrajcelično proteolize HIF. Tako je prisotnost kisika uniči HIF. Kisik stradanje celic ali odsotnosti VHL genskih funkcionalnih izdelkov pospešuje kopičenje HIE HIF so transkripcijski faktorji, ki inducirajo sintezo vaskularnega endotelijskega rastnega faktorja (ERS) eritropoetin eritropoetinskega receptorja, GLUT-1 in trombocitni rastni faktor-B. Vsi ti proteini so prilagojene celice do hipoksije, vendar je njihova številčnost je verjel, da prispeva k razvoju tumorjev.
Bolniki z razvojno BGL hemangioblastomas, cist in raka ledvic jasno, ponavadi zahteva somatsko mutacijo preostali divjega tipa alel gena VHL (izgubo heterozigotnosti) ali hypermethylation svojega promotorja (tako imenovani "drugi hit"). Z drugimi besedami, ki zadostuje za razvoj teh kopičenja tumorji HIF pojavi le, če obe aleli okvar VHL gen. Feokromocitom je BGL pri bolnikih s tipom 2 razvijajo v normalnem stanju drugega alela gena. Zato je bilo predlagano, da nekateri missense mutacije v tem genu razvoju vzrok za feokromocitom, uveljavljajo svoj vpliv drugačen mehanizem, in sicer - s prekinitvijo vezavo sintetiziranega proteina fibronektin.
Dedne (zarodne) VHL genske mutacije so na voljo v večini družin z BGL. Približno 60% teh mutacij so izgube funkcije (30% okrnjene proteini sintetizirajo, in 30% - so glavne delitev gen), ki povzroča BGL tipa 1. Približno 40% bolnikov so nosilci missense mutacij, ki vodijo do aminokislinske substitucije kisline v proteinu, ki se sintetizira. V teh primerih se razvije BGL diabetes 2.
} {Modul direkt4
Pri 53% od 36 bolnikov z LGL opazili v Franciji, so bili prvi odkrivanje tumorjev samo feokromocitoma, ki se pogosto pojavlja v zgodnjem otroštvu in v 42% primerov so dvostranski. Hkrati je bilo ugotovljeno 11% bolnikov paraganglioma. Maligni feokromocitoma pojavila pri treh od 36 bolnyh- 18% od feokromocitom bila edina manifestacija bolezni. Približno 9% bolnikov z enostransko feokromocitom (izhaja sporadična) izkazal prevoznikom zarodne VHL genskih mutacij. V nekaterih delih Evrope je število bolnikov je 20%, kar je ustrezalo "ustanovitelja učinek" (zamenjava Tir98Gis v VHL genskega produkta, tako imenovani "Schwarzwald mutacije" skupne družine nemškega porekla).
Feokromocitoma na BGL proizvajajo samo noradrenalin in njegov presnovek - Normetanephrine katerih raven v plazmi v takšnih primerih običajno poveča. Zato BGL pri bolnikih z tipa 2 (missense genske mutacije VHL) treba redno normetanephrine določimo koncentracijo v plazmi.
Največja nevarnost za življenje bolnikov z LGL, je jasno, karcinom ledvičnih celic. Vsak trdna ledvični tumor, CT trebuha diagnosticirana, ki jih je treba odstraniti. Tudi ciste na ledvicah teh bolnikov se štejejo za predrakave in jih bolje odstraniti. Če takih tvorb vsakih 6 mesecev, je potrebno izvesti visoke ločljivosti CT zaznati znake malignosti: povečanje velikosti, ciste neravne stene in predelne stene nastop v njih. Prevozniki VHL genske mutacije treba skrbno spremljati.
- BGL tipa 1. Na vrsti BGL 1 feokromocitom ne razvijajo. VHL mutacije gena običajno vodijo do izgube njegove funkcije (črtanje premakne bralni okvir ali sintezo okrnjenih proteinov).
- BGL diabetesa 2. V teh primerih je zaradi missense mutacij v VHL gena, feokromocitom običajno razvije. diabetesa BGL 2 je razdeljen na 3 podtipov:
- BGL tipa 2A (hemangioblastoma in feokromocitom, vendar nizko tveganje za razvoj raka ledvic jasnih);
- BGL tip 2B (hemangioblastoma, feokromocitom in visoko tveganje za razvoj raka ledvic jasnih);
- Tip BGL 2C (brez hemangioblastomas feokromocitom ali raka ledvic).
Neirofibromatoz tipa 1 (Recklinghausen bolezen)
Feokromocitom značilnost 0,1-5,7% bolnikov Recklinghausen nevrofibromatoza s tipom 1 (NF-1). Večina teh tumorjev ne zazna v času življenja, saj je obdukcija pri bolnikih z NF-1, njihovo frekvenco 3,3-13%. So podobni sporadične feokromocitom: pri odkritih 84% primerov en tumorja pri eni nadledvične žleze, 10% - obojestransko tumorja pri 6% bolnikov ugotovljenih extraadrenal paraganglioma in 12% - metastaze ali invazije okoliških tkivih.
Feokromocitom najdemo v 20-50% bolnikov z NF 1 z arterijsko hipertenzijo. Zato je v vseh teh primerih je treba prevzeti prisotnost feokromocitom. Primerno raziskave bi bilo primerno letno splošno vsi bolniki NF-1, in v časovnih presledkih, za merjenje krvnega tlaka in ugotovite prisotnost simptomov, značilnih za feokromocitom (glavobol, znojenje, palpitacije). Iskanje feokromocitom pri bolnikih z NF-1, mora biti tudi pred večjimi operacijami in nosečnosti.
Feokromocitoma z NF-1, se lahko razvije pri katerikoli starosti - od otroštva do starosti- povprečna starost bolnikov ob diagnozi je 42 let. Ti tumorji pogosto dosežejo velike velikosti. Presenetljivo je, da mnogi bolniki, kljub povečanim izločanjem kateholaminov in krvnega tlaka ostalo normalno. Ko SF-1 je pogosto opaziti koarktacija aorte in ledvic displazije arterij, ki lahko povzročajo hipertenzijo, feokromocitom simulaciji.
Osnova Recklinghausen mutacija bolezen leži NF-1 (zaviralnih), ki se nahaja na kromosomu 17 (odsek 17q11.2) in kodira neyrofibromin (protein po 2818 aminokislinskih ostankov), ki inhibira proteini RAS. Neyrofibromina odsotnost vodi v razvoj tumorjev. NF-1 je avtosomno dominantna motnja (čeprav se zdi, da sporadično 50% primerov) - njegova frekvenca je okoli 300-400 na 1.000 prebivalcev 000. Diagnoza se običajno določi v otroštvu ali adolescenci na podlagi kliniki, čeprav je mogoče in genetske analize.
Otrok SF-1 se običajno manifestira gliomov za vidnega živca, ki vodi do slabovidnost, in kot najstnik - plexiform neurofibromas. Bolniki prikazani subkutane neurofibromas in schwannomi korenine lobanjskih in spinalnih živcev. Pogosto obstajajo nepravilnosti skeleta. Hipotalamusa hamartomov lahko povzroči prezgodnji spolni razvoj. Včasih so iris hamartomov (Lisch vozlički). Povečano tveganje za razvoj drugih tumorjev (zlasti malignih tumorjev perifernih živcev membran) in levkemij (zlasti juvenilni kronične mieloične levkemije). Značilna pege pod pazduho in gube na koži. Obstaja veliko lise barvi kave z mlekom, števila in velikosti, ki povečujejo s starostjo. Večina bolnikov s več kot šest Madeže z gladkimi robovi in 1,5 cm v premeru.
Prevozniki NF-1 genskih mutacij treba skrbno spremljati.
Družina sindromih paraganglia / feokromocitom: genskih mutacij suktsinatdegidrogenaznogo kompleks
Člani nekaterih družinah pogosto razvijejo Multicentrična paragangliomas iz glave in vratu, simpatičnega paraganglioma in nadledvične feokromocitom. Osnova teh sindromov so trije od štirih mutacij genov, ki kodirajo mitohondrijske kompleks II, ki obsega SDGV in SDGS SDGD. V nizozemski družini s sindromom odkrili mutacijo gena, ki se nahaja na kromosomu 11.
Pri vseh bolnikih z mutacijami SDGV geni SDGS in SDGD (očetovi alelov), nagnjenost k razvoju paraganglia podedovanih v avtosomnem prevladujoči način. Ko mutacije genov razvijajo SDGD SDGV in tudi nadledvične žleze feokromocitoma. Pri osebah z genskih mutacij pride SDGD paragangliomas le v primerih, ko je mutant gen podedovali od svojega očeta. Nosilci gena mutacije zarodnih LDH je približno 12% bolnikov s temi sindromi.
Frekvenca paraganglia glave / vratu je 10-33 primerov na 1 milijon prebivalcev. V seriji opazovanj, ki je vključevala 34 bolnikov s takšnimi tumorji, so kalčki LDH genske mutacije na voljo v 41% primerov. Med bolniki s temi mutacijami v 79% primerov je bilo genskih mutacij SDGD in ostalo - genskih mutacij SDGV.
Kot smo že omenili, so vsi ti sindromi povzročijo mutacije v jedrskih genov, ki kodirajo tri od štirih podenot, ki tvorijo mitohondrijske kompleks II (SDH), ki je v ciklu Krebs oksidira sukcinat na fumarat. Oligomerni LDH vključuje SDGA (flavoprotein z molekulsko maso 70 kDa) SDGV (železo žvepla proteina, 30 kDa), SDGS (podenota citokrom b, 15 kDa) in SDGD (druga podenota citokroma b, 12 kDa). Komponente LDH gen, ki obsega b (SDGS in SDGD) lokaliziran v mitohondrijski membrani, ki je vezana podenoto SDGA SDGV ki veriga transporta elektronov pri prenosu elektronov koencim Q (ubikinon). SDG je potrebna za cikel in energije citronske kisline v aerobnih pogojih. Genetske napake SDG krši mitohondrijske funkcije, ki povzročajo hipoksičnih celice. To povečuje izločanje vaskularnega endotelijskega rastnega faktorja (ERS) zahteva za napredovanje tumorja. (Embrionalne SDGA mutacije povzročijo razvoj chromaffin telesa ni, in ali bolezni - smrtna bolezen nevrodegenerativna otroštvo, ki ga mitohondrijske bolezni, povzroča).
- Mutacije SDGD gen. SDGD gen lokalizirana na kromosom 11 (odsekih Q23). Mutacije tega gena so našli pri večini bolnikov z družinsko paraganglioma. V družinah z nagnjenost do tovrstnih mutacij razvojnih paraganglia feokromocitom in vpisati le v primeru dedovanja mutant gena od očeta ( "mater genomske odtis"). Moški Otroci, ki so prejeli mutirani gen iz SDGD matere, ne zbolijo, vendar pa se lahko prenese na gen (in s tem predispozicije za razvoj chromaffin telesu) na svoje potomstvo. Ženske, ki so prejeli mutirani gen od očeta, paragangliomas razvija, vendar svoje otroke - ne. Pri osebah z reproduktivnih mutacije gena SDGD zlasti pogosto pojavijo tumorji parasimpatično ganglije (embrionalni povezana z simpatičnega chromaffin telesu) v glavi in vratu (angioneuroma). Ker ti paraganglioma skoraj ne izločajo kateholamine, ki jih imenujemo "non-chromaffin". Angioneuroma razvoj, običajno zaspano Glomus na razcepu karotidne arterije (chemodectoma) ali na dnu lobanje in sta eno ali več, enojno in dvojno. Vsi taki bolniki potrebujejo skrbno študijo območja vratu, skeniranje in redno vseživljenjsko spremljanje. Približno 74% bolnikov z paraganglioma mutacije SDGD gena povzroča Zaznali Multicentrična tumorjev, in 50% bolnikov z navidezno izoliranih feokromocitomnih in reproduktivnih mutacij v genu so skriti SDGD paraganglioma. Tako je v vseh teh primerih je treba iskati druge paraganglioma uporabo MRI in / ali pozitronska emisijska tomografija (PET). V družinah z nizozemskega porekla odkrita dva "ustanovitelj mutacijo" gena SDGD: Ley95Pro in Asp92Tir. "Mutacija ustanovitelja" (Gln109H) najdemo tudi v družinah italijanskega porekla. Verjetnost razvoja chromaffin telo (penetrantnih) v nosilci mutirane gena SDGD starševske starostjo narašča od 33% v starosti od 30 do 83% v 60 letih. Paraganglioma, z mutacijami v genu SDGO povzročil redko rakav. Vendar pa lahko materničnega vratu paraganglioma metastaze na regionalne bezgavke, pljuča in kosti- včasih mnogo let ne pokažejo klinično. Morda razvoj chromaffin telesa in na drugih mestih, kot tudi feokromocitom, ki zahteva vseživljenjsko spremljanje teh bolnikov.
- Mutacije SDGV gen. SDGV gen je preslikan v kromosomu 1 (odsek 1r36). Mutacije tega gena so povezani z paraganglioma manj kot gen SDGD mutacije. Tumorji v teh primerih lahko kjerkoli razvije na parasimpatičnega in simpatičnega sistemov - od vratu do medenice, kakor tudi v nadledvičnih žlezah (feokromocitomnih), vendar zaspan glomus v vratu prihaja manj pogosto kot pri mutacij SDGD gena. Na embrionalnih genske mutacije SDGV paraganglia metastaze v času diagnoze zazna pogosteje (35% primerov) kot v semenskih SDGD genskih mutacij. Izračunana SDGV penetrantnih mutirane gena je približno 31%. Takšni bolniki so bolj verjetno, da razvijejo druge raka. Od 53 bolnikov z mutacijami v tem genu imajo dve jasno karcinom ledvičnih celic je bila odkrita in ena - papilarni rak ščitnice.
- Mutacije SDGS gen. SDGS gen lociran na kromosomu 1 (odsekih Q2). Ko mutacije tega gena paraganglioma zaznali le v eno evropsko družino, in vsi bolniki so bili zunaj nadledvične žleze. Tako je treba vse bolnike z paraganglioma in feokromocitom prevzemajo prisotnosti semenskih mutacij genov, ki kodirajo SDH kompleks. Genetska analiza je zlasti navedeno v primerih paraganglia vratu (v kateri so seme in mutacije več kot 15% bolnikov), ali multifokalna paraganglia feokromocitom, chromaffin organ ali družinsko anamnezo feokromocitom in bolniki paraganglia nizozemskega izvora.
Drugi genetski sindromi, vključno feokromocitoma
- Carney triade. Multicentrična paragangliomas pojavi pri bolnikih z Carney triade (ne sme zamenjati z kompleksa Carney!). Ta sindrom se navadno pojavi pri ženskah, mlajših od 40 let in vključuje paraganglioma skrito leyomiosar-komo želodec in pljuč chondroma.
- Beckwith-Wiedemann sindrom. Feokromocitom (vključno z obeh strani) so se pri bolnikih z BWS opazili. Bolniki so opazili in druge kršitve, zlasti - hipoglikemija novorojenčka, popkovna kila, hernia popkovini, macroglossia in gigantizem, pa tudi povečano tveganje malignih tumorjev.
 Povezava med malformacije in tumorjev. Dedni vzroki otroških tumorjev
Povezava med malformacije in tumorjev. Dedni vzroki otroških tumorjev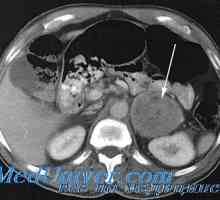 Feokromocitoma: klinični, patološki mehanizmi razvoja
Feokromocitoma: klinični, patološki mehanizmi razvoja Simpatogoniomy in feohromoblastomy. organi spreminjajo s feokromocitom
Simpatogoniomy in feohromoblastomy. organi spreminjajo s feokromocitom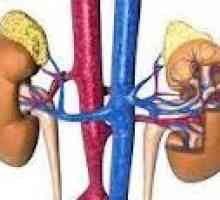 Vzroki in patogeneza feokromocitom
Vzroki in patogeneza feokromocitom Simptomi feokromocitom pri otrocih
Simptomi feokromocitom pri otrocih Simptomi in znaki feokromocitom
Simptomi in znaki feokromocitom Zdravljenje feokromocitom
Zdravljenje feokromocitom Diagnostika in analize s feokromocitom
Diagnostika in analize s feokromocitom Adrenalne feokromocitoma - maligna tumorja, bolezni, foto kliniki, oznaka po ICD-10
Adrenalne feokromocitoma - maligna tumorja, bolezni, foto kliniki, oznaka po ICD-10 Feokromocitoma simptomi, diagnosticiranje in zdravljenje feokromocitom
Feokromocitoma simptomi, diagnosticiranje in zdravljenje feokromocitom- Nadledvične hipertenzija
- Feokromocitom, bolezen, ki jo je benigni ali maligni tumor nadledvične chromaffin tkiva ali…
- Razlikovati benigne in zlokachestvennyeref = "des204.htm"> nadledvične tumorjev, ki…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Onkologiya-
 Feokromocitoma in paraganglioma: simptomi, znaki, zdravljenje, vzroki
Feokromocitoma in paraganglioma: simptomi, znaki, zdravljenje, vzroki Medularni rak ščitnice
Medularni rak ščitnice Sindromom multiple endokrine neoplazije tipa II (Maine 2): vzroki, simptomi, zdravljenje, simptomi
Sindromom multiple endokrine neoplazije tipa II (Maine 2): vzroki, simptomi, zdravljenje, simptomi Stanja, povezana s prekomernim izločanjem hormonov nadledvične žleze
Stanja, povezana s prekomernim izločanjem hormonov nadledvične žleze Bolezen s številnimi lezij endokrinih žlez
Bolezen s številnimi lezij endokrinih žlez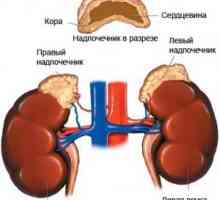 Tumorji skorje nadledvične žleze: simptomi, zdravljenje, vzroki, diagnoza, simptomi
Tumorji skorje nadledvične žleze: simptomi, zdravljenje, vzroki, diagnoza, simptomi