Ogljikovi hidrati metabolizem pri dojenčkih

Vsebina
- Video: kršitev presnove ogljikovih hidratov pri otrocih
- Bolezni glikogena
- Video: film samostana. enostavni ogljikovi hidrati. visoka glikemični indeks
- Motnje presnove fruktoze
- Motnje piruvat presnove
- Video: patofiziologije presnove ogljikovih hidratov (predavanje). t.n. alhendi. del 2/2
- Video: prirojenimi napakami v metabolizmu ogljikovih hidratov. klinični znaki, diagnostika, zdravljenje
- Druge motnje presnove ogljikovih hidratov
Galaktozemije - avtosomno recesivna bolezen pojavi pri 1 otrok do 60 000 živorojenih otrok.
V klasičnem, pogostejšega obliki pomanjkanja galaktoza-1-fosfaturidiltransferazy ki vodi do kopičenja galaktoza, galaktoza-1-fosfat in galaktiola tkiva. s pomanjkanjem galaktozemije galaktokinaza ali epimeraze manj pogosto povzroča. Na sprejemu v laktoze pojavijo novorojenčka bruhanje hrane, driska, hiperbilirubinemije, hepatosplenomegalija, ledvično tubularno disfunkcijo, z odpovedjo jeter, sive mrene. Galaktoze v krvi bolnih otrok.
Diagnoza je potrjena z oceno aktivnosti galaktoza-1-fosfatgransferazy v rdečih krvnih celic, ki bo res, če se ne pred rdečo transfuzij krvnih celic.
Morfološke spremembe vključujejo izgovarja s postopnim jetrna steatoza psevdoatsinarnoy preureditev strukture lobules proliferacijo žolčnih vodov, holestaze goriščni nekroze z rezultatom v cirozo.
Poleg tega otočnih celic hiperplazijo, vakuolacijo nephrothelial nespecifično ishemične možganske poškodbe v obliki smrti nevronov, glioze, edem.
Morfološke spremembe na dedno intoleranco za fruktozo in tirozinemije so podobni (glej. Spodaj).
Kršitve presnove fruktoze. Predstavljajo bolezni z avtosomno recesivno način dedovanja, očitno v novorojenčka roka za sprejem s hrano. Se kaže kot bruhanje, slabost, hepatomegaliji, odpovedi jeter, hipoglikemije, laktacidoze. Steatoze, holestaze portala fibroza in proliferacijo cholangioles s pretvorbo v cirozo.
Glycogenoses - skupina podedovanih motenj metabolizma ogljikovih hidratov, ki je posledica mutacij v več genov, ki kodirajo encime, ki urejajo sintezo in razgradnjo glikogena v enostavnih sladkorjev in normalno ali nenormalno kopičenje glikogena v celicah mnogih tkivih. Razvrstitev temelji na napako določenega encima. Deduje avtosomno recesivno način, razen glikogenoza tipa VIII, spol vezana.
Raznolikost oblik bolezni skladiščenja glikogena (tip 14) odraža vlogo v glikogen presnovi številnih genov.
Večina bolnikov ima sistemskih manifestacij opazili v nekaterih oblikah okvare posameznih organov. Značilna simptomi miopatije, pa tudi epizod hipoglikemije, kardiomegalija razvoj.
Video: Kršitev presnove ogljikovih hidratov pri otrocih
bolezni glikogena
bolezni glikogena (GSDs) zaradi nezadostne encimov, ki sodelujejo pri sintezi ali pomanjkljivostmi uničenja glikogena- lahko pojavijo v jetrih, mišicah in povzroči hipoglikemijo ali odlaganje nenormalnih količin ali vrst nenormalne glikogena v tkivih.
Video: Film samostana. Enostavni ogljikovi hidrati. Visoka glikemični indeks
Dedovanje GSDs avtosomno recesivna razen vrst GSD VIII / IX, X-vezana. Pojavnost je ocenjena na približno 1/25 000 rojstev, medtem ko se lahko podcenjevati, saj so lahki srcno oblike niso vedno postavljena diagnoza.
Starost nastopom, klinične manifestacije in resnosti odvisna od vrste.
Diagnoza je potrjena z znatnim zmanjšanjem encimske aktivnosti v jetrih (tipov I, III, VI in VIII / IX), mišice (tip llb, III, VII in VIII / IX), kožne fibroblastov (vrste Ha in IV) ali eritrocitov (tipa VII) odsotnost ali povečajo aktivnost venskega laktat / ishemije podlakti (tipa v in VII). Prognoza in zdravljenje se razlikujejo glede na vrsto, vendar je zdravljenje pogosto vključuje prehranske dodatke s koruznim škrobom zagotoviti stalen vir glukoze v oblikah jeter iz GSD, in vaje za preprečevanje oblike mišic.
Napake v glikoliza (redko), lahko povzroči sindrom podobno GSDs. Fosfoglicerat pomanjkljivost kinaze, fosfoglicerat mutazo in laktat miopatija posnema tipa GSD V in VII-primanjkljaji prevoz glukoze proteinski 2 (jabolčnik Fanconijeve - Bickel) posnema GSD hepatopathy druge vrste (npr 1.111, IV, VI).
Motnje presnove fruktoze
Pomanjkanje presnovnega encima fruktoza, lahko asimptomatičnih ali povzroči hipoglikemijo.
Fruktoza je monosaharid prisoten v visokih koncentracijah v sadju in medu in ki je del saharoze in sorbitola.
Pomanjkanje fruktoza-1-fosfat aldolaze (aldolaza B). To pomanjkljivost je klinični sindrom, dedno fruktoza nestrpnosti. Dedovanje se je avtosomno retsessivnoe- incidenca ocenjena na 1/20 000 rojstev. Dojenčki ostanejo zdravi pa ne porabijo fruktoze. Dolgotrajna uporaba lahko povzroči cirozo, duševnega poslabšanja in ledvično tubularno acidozo proksimalno odsotnosti fosfata v urinu in glukozo.
Diagnoza temelji na simptome predlagamo glede na nedavno vnosa fruktoze in potrditev encimske analize jetrnega tkiva biopsiji ali z indukcijo hipoglikemija intravenozno infuzijo fruktozo v količini 200 mg / kg.
Kratkoročni zdravljenje je sprejemanje glukoze gipoglikemii- dolgotrajno zdravljenje je odstranitev porabe fruktoze, saharoze in sorbitola. Mnogi bolniki razvoj naravne fruktoze nestrpnost hrano. Pri zdravljenju je prognoza je odlična.
fructokinase primanjkljaj. Ta pomanjkljivost povzroči benigno povečanje koncentracije v krvi in urinu fruktoze (benigna fructosuria). Dedovanje je avtosomno retsessivnoe- incidenca je približno 1/130 000 rojstev.
Status asimptomatski in po naključju odkrijejo pri odkrivanju sečil neglyukozovosstanavlivayuschego snovi.
Pomanjkanje fruktoza-1, 6-bisfosfatazni. To pomanjkljivost ogroža glukoneogenezo in vodi do tešče hipoglikemija, ketoza in acidozo. Pomanjkanje je lahko usodna za dojenčke. Dedovanje Avtonomno retsessivnoe- pogostnost ni znana. Obstajajo epizode povišane telesne temperature.
Nujna Obdelava vključuje oralno ali intravensko dajanje glukoze. Toleranca do lakote običajno povečuje s starostjo.
Motnje piruvat presnove
Nezmožnost, da prebavi piruvat vodi v razvoj laktacidoze in različnimi motnjami centralnega živčnega sistema.
Video: patofiziologije presnove ogljikovih hidratov (predavanje). T.N. Alhendi. del 2/2
Piruvat je pomemben substrat metabolizma ogljikovih hidratov.
Pomanjkanje piruvat dehidrogenaze. Piru vatdegidrogenaza je multiencimskega kompleks odgovorna za tvorbo acetil-CoA iz piruvata za cikel Krebs. Pomanjkanje vodi do povečane stopnje piruvata in s tem povečati raven mlečne kisline. -X vezano dedovanje ali avto-kromosomskih recesivna.
Video: prirojenimi napakami v metabolizmu ogljikovih hidratov. Klinični znaki, diagnostika, zdravljenje
Klinične manifestacije razlikujejo po resnosti, vsebujejo pa laktacidoze.
Diagnozo potrdimo z encimom testnem aktivnost analize fibroblastov kože DNA teh dveh metod.
Jasno je, da ni učinkovito zdravljenje, medtem ko je bila dieta z nizko vsebnostjo ogljikovih hidratov ali ketogenic prehrane in prehranskih dopolnil tiamina koristno za nekatere bolnike.
Pomanjkanje piruvat karboksilaza. Pomanjkanje lahko primarni ali sekundarni na pomanjkljivost golokarboksilazy Sintetaze biotin ali biotinidazy- avtosomno recesivno dedovanje, in obe različici vodi do laktacidoze.
Pojavnost primarnega primanjkljaja <1/250 000 родов, но может быть выше у некоторых американских индейских народов. Задержка психомоторного развития с эпилептическими припадками и спастичность являются основными клиническими проявлениями. Нарушения лабораторных показателей включают гипераммониемию- молочный ацидоз, кетоацидоз.
Klinični znaki sekundarnega pomanjkanja so podobni in vključujejo nenapredovanja, epileptične napade in druge organske aciduria.
Učinkovito zdravljenje ne obstaja, vendar pa so nekateri bolniki s primarnim primanjkljajem in vse osebe, ki imajo povprečno resnost bolezni, je treba dodati biotin 5-20 mg peroralno enkrat na dan.
Druge motnje presnove ogljikovih hidratov
Pomanjkanje fosfoenolpiruvat karboksikinaze (261680) daje glukoneogenezo in vodi do simptomov in znakov, ki so podobni jeter obliki glikogenoza, vendar brez kopičenja glikogena v jetrih.
Druge pomanjkljivosti vključujejo okvari glikolitičen encime ali encime iz poti pentoza-fosfata. Tipični primeri so pomanjkanje piruvat kinaze in glukoza-6-fosfat dehidrogenaze (G6PD), kar lahko vodi do razvoja hemolitične anemije. Wernicke sindrom - Korsakoffovo povzročil delnem izpadu transketolase poti pentoza-fosfata je encim, ki zahteva tiamin in kot kofaktor.
 Laktofiltrum driska
Laktofiltrum driska Poslezheltushnaya encefalopatija. Microspherocytosis dedna bolezen Minkowski-Chauffard
Poslezheltushnaya encefalopatija. Microspherocytosis dedna bolezen Minkowski-Chauffard Razvrstitev neonatalne zlatenice. izraelsa sindrom
Razvrstitev neonatalne zlatenice. izraelsa sindrom Kombinirana imunska z fermentopathy. nezelofa sindrom ali alymphocytosis
Kombinirana imunska z fermentopathy. nezelofa sindrom ali alymphocytosis Smrt intrauterini plodu z maceracijo. Simptomi hemolitično bolezen novorojenčka
Smrt intrauterini plodu z maceracijo. Simptomi hemolitično bolezen novorojenčka Galaktozemije
Galaktozemije Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji
Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji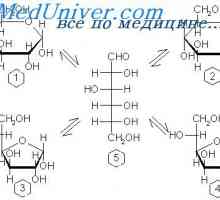 Fiziologija presnove glukoze. Transport glukoze preko celične membrane
Fiziologija presnove glukoze. Transport glukoze preko celične membrane Avtosomno recesivna policističnih ledvic pri otrocih. Diagnoza in zdravljenje
Avtosomno recesivna policističnih ledvic pri otrocih. Diagnoza in zdravljenje Neonatalna zlatenica s metaboličnih motenj
Neonatalna zlatenica s metaboličnih motenj Hiperbilirubinemija funkcionalno (benigna hiperbilirubinemija, zlatenica funkcionalno) skupina…
Hiperbilirubinemija funkcionalno (benigna hiperbilirubinemija, zlatenica funkcionalno) skupina…- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Terapija, bolezni prebavnega sistema
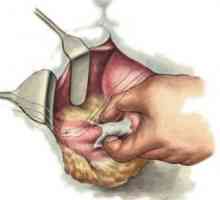 Žolčevoda atrezija
Žolčevoda atrezija Izmenjava prekrški kovine
Izmenjava prekrški kovine Prirojeno in dedno tubulopatiji pri otrocih
Prirojeno in dedno tubulopatiji pri otrocih Metaboličnih motenj presnove sečne kisline in kovine
Metaboličnih motenj presnove sečne kisline in kovine Ciroza jeter pri otrocih simptomi
Ciroza jeter pri otrocih simptomi