Lizozomalnih bolezni pri otrocih

Vsebina
Veliko več kot 40 opisane dedne bolezni s poškodbami lizosomalnih encimov označen z boleznijo jeter.
To velja predvsem za Gaucherjeve bolezni, Niemann - Pick, Wolman, število mukopolisaharidoze, oligosaharidozov, metachromatic leukodystrophy, Farber bolezni, Fabryja, gangliosidosis tipa I.
Gaucherjeva bolezen. Obstaja povečana jetra. Histološko določi Gaucherjeve celice, ki so razširjeni Kupfferjevih celice z ekscentrično nahaja jedrom in citoplazmo značilnost ima obliko "zmečkan tissue papirja." Pri razporeditvi skupin Gaucherjevo celic v jetrih lobule in portalnih trakta so lahko označene fibroze. Lahko razvije ciroza.
Bolezen Niemann - Pickovo. S sodelovanjem v patološkem postopku jeter da postane razširjeni, bledo. Mikroskopsko: je struktura lobules lomljeno, fibroza ni značilno. Kupfferjeve celice povečala, pena so vakuoliziranega citoplazmo. Podobne spremembe so v hepatocitov opazili. V celicah, kopičenje ceroidna, holesterola in fosfolipidov. Elektronska mikroskopija je pokazala značilno mielinsko lik, sestavljen iz koncentričnih ali vzporednih osmiophilic plošč. V kliničnem aktivnosti izvedbenem bolezen sfingomielinaze z normalnim morfološki izgled je mogoče opaziti v jetrih in levkocitov, take neonatalne hepatitisa.
Wolman bolezen. Jetra se poveča, pobarvana v rumeno. Histološko steatoze pojavi ob prisotnosti velikega števila penastih celic, ki so hepatocitov Kupfferjeve celice, ki vsebujejo velike količine holesterola in maščob. Holesterol je najbolje pokazala s pregledom odsekov jeter tkiva zamrznjenih polarizirane svetlobe.
Mukopolisharidoze. poškodbe jeter zaradi kopičenja v celicah in medceličnem snovi in kislih glikozaminoglikani povzročila istočasno gangliozidom. Obstaja povečana jetra. Histološko kaže vakuolizacijo hepatocitov in Kupfferjeve celice in v manjši meri žolčevoda epitelija. Lahko sčasoma razvijejo fibrozo jeter.
Mucolipidosis. Jetra se poveča, pobarvana v rumeno. Histološko steatoze pojavi ob prisotnosti velikega števila penastih celic, ki so hepatocitov Kupfferjeve celice, ki vsebujejo velike količine holesterola in maščob. Holesterol je najbolje pokazala s pregledom odsekov jeter tkiva zamrznjenih polarizirane svetlobe.
Oligosaharidozy. Jetra so vključeni v proces v vseh oblikah bolezni. Obstaja hepatomegalija s vacuolization v citoplazmi hepatocitov in Kupfferjevih celic. Vakuole so različnih velikosti, se lahko zberejo v precej veliki. Vakuolacijo lahko pojavi tudi v endotelijskih celicah in žolčevoda epitelija.
Metachromatic leukodystrophy. V makrofagov v portala trakta jeter določimo metachromatic granul, in jih je mogoče zaznati tudi v Kupfferjevih celice in fibroblaste. Žolčniku lahko določimo papilarni izrastki sluznico prisotnosti penastih celic v subepitelno coni.
Farber bolezen. Obstaja povečana jetra. Akumulacija ceramid in gangliozidov pojavi pri jetrnih vacuolization v citoplazmo, pri čemer se pridobijo videz penast. Poleg tega se razvija reaktivno granulomatozno proces z izidom fibroze. Lipogranulemy razpršene jetrno tkivo, sestavljeno iz limfocitov, makrofagov, večjedrne celic velikank.
Fabry bolezni. Kot pri drugih organov, kopičenje jetra globotriaosiltseramida pojavlja predvsem v vaskularnih endotelijskih celicah.
Gangliosidosis. V oblikah bolezni vključuje jetra pojavi hepatomegalije. Svetlobna mikroskopija je označena vacuolization v citoplazmi hepatocitov in Kupfferjevih celic, ki jim daje penasto videz. Elektronska mikroskopija povečana pregledne odločno lizosome, ki vsebujejo granulatom mrežo.
bolezni lizozomalnih shranjevalnih
Lizosomski encimi razgradijo makromolekule ali same celice (npr ko recikliranih komponente strukturne celice) ali ujetih od zunaj. Dedne napake ali pomanjkljivosti lizosomalnih encimov (lizosomatskega ali druge sestavine) lahko vodi do kopičenja nerazgrajenost metabolitov. S prisotnostjo številnih posameznih bolezni pomanjkljivosti shranjevanje so običajno razvrščene po katerem biokemično akumuliranega metabolit.
Podskupine so:
- mukopolisaharidoza,
- sfingolipidoz (lipidoses)
- Mucolipidosis.
To je najbolj pomembno in mukopolisaharidoza sfingolipidoz. Tip 2 glikogen bolezen shranjevanje je motnja shranjevanje lizozomalnih, vendar je večina glikogena - št.
Ker retikuloendotelijskega celic (npr vranica) bogatih lizosome, retikuloendotelijskega tkiva nekaterih bolezni shranjevanje lizosomski poškodovana, vendar običajno tkiva najbogatejši substrat trpijo vsego.Takim močnejši način, možgani, ki je bogata z gangliozidi, zlasti trpijo gangliosidosis, ker mukopolisaharidoza prizadene veliko tkiva, saj so prisotne v telesu mukopolisaharidov.
Mukopolisharidoze (MPS). Mukopolisharidoze - dedno pomanjkanje encimov, ki sodelujejo pri razgradnji glikozaminoglikanove. Glikozaminoglikani (prej znan kot mukopolisaharidov) so konvencionalne in polisaharidi celične površine in ekstracelularnega matriksa strukture. Pomanjkanje encima, ki preprečuje uničenje glikozaminoglikanov, povzroči kopičenje glikozaminoglikan fragmentov v lizosomih in povzročila obsežne spremembe v kosti, mehko tkivo in centralni živčni sistem. Dedovanje običajno avtosomno recesivna (razen mukopolisaharidoza tipa II).
Starost, klinične manifestacije in resnost se razlikujejo glede na vrsto.
Splošni znaki vključujejo grobe poteze obraza, upočasni razvoj živcev in nazadovanje, skupne kontraktur, organomegalijo, grobo lase, progresivno odpoved dihal, srčne okvare, spremembe okostja in subluxation od vratnih vretenc.
Diagnozo je predlagal anamnezo, podatke o fizični pregled, nepravilnosti v kosteh (npr multipla dysostosis), so bili med študijo skeleta odkrije in zvišane skupnega in frakcionirani glikozaminoglikani. Diagnozo potrdimo z encimom testom v kulturi fibroblastov (pred rojstvom) ali perifernih levkocitih (po rojstvu). Dodatno testiranje se izvaja za nadzor gibanja organ specifičnega (npr pomanjkljivosti ehokardiografija ventil, avdiometrija za odkrivanje sprememb v obravnavi).
Zdravljenje mukopolisaharidoze tipa I (Hurler bolezen) vključuje polnjenje encimsko -L-iduronidazy, ki učinkovito ustavi napredovanje in obnovi vseh zapletov ne-CNS bolezni. Presaditvi hematopoetskih matičnih celic (HSCs) se je izkazala za učinkovito v zgodnjih raziskavah, vendar neučinkovit pri bolezni CŽS.
Sfingolipidoz. Sfingolipidi - normalni lipidne sestavine celičnih membran, se kopiči v lizosomih in povzročila obsežne spremembe v nevronih, kosti in druge spremembe pri encimski defekt preprečuje njihovo uničenje. Čeprav je incidenca je nizka, nekatere oblike nosilno frekvenco je visoka. Gaucherjeva bolezen je najpogostejši fingolipidozom.
Gaucherjeva bolezen
Gaucherjeva bolezen je sfingolipidozom, razvoju zaradi pomanjkanja glukocerebrozidaza, ki vodi do kopičenja glukocerebrozida in sorodnih spojin. Simptomi in znaki se razlikujejo ottipa.
Tip I (neneyropatichesky) je najbolj razširjena (90% vseh bolnikov). Preostala encimska aktivnost je maksimalna. Aškenazi so najbolj risku- 1/12 so nosilci. Starost začetka bolezni se giblje od 2 leti do konca odraslo življenje. Simptomi so hepatosplenomegalija, bolezni kosti, zaostajanje v rasti, zapoznelo puberteto, odrgnine in pinguecula. Nosni krvavitve in modrice, ki izhaja trombocitopenijo so skupne manifestacije. Radiografija prikazuje najpomembnejše koncih dolgih kosti (kolboobraznaya erlenmajerica deformacije) in skorje redčenje.
Tipa II (akutna nevropatska). Pojav bolezni v obdobju otroštva.
Tipa III (subakutni nevropatska) o obolevnosti, encimske aktivnosti in klinične resnost nahaja med vrstami I in II. Bolezen se pojavi kadarkoli v času otroštva. Klinični znaki so odvisni od podtipa in vključujejo progresivno demenco in ataksijo (Ilia).
diagnostika
- Določitev encimske aktivnosti.
Diagnoza temelji na analizi levkocitov encimov. Prevozniki prepoznavanje in razlikovanje vrst z analizo mutacij. Čeprav biopsija ni potrebna Gaucherjeve celice - lipidne injekcijskih makrofagov v jetrih, vranici in kostnem mozgu, imajo obliko tanek papir zmečkan - so diagnostični.
zdravljenje
- V tipov I in III: dolivanje encim ali rekombinanten placente glukocerebrozidaza.
- Včasih Miglustat, splenektomija matičnih presaditev celic.
Bolniki, ki prejemajo nadomestno encimsko, zahtevajo redno spremljanje koncentracije hemoglobina in trombocitov, redno ocenjevanje vranice in države prostornine jeter, CT ali MRI, pa tudi redno ocenjevanje stanja bolezni kosti v skeletni raziskavi, dvojno energijo X-ray ab sorbtsiometrii ali MRI.
Splenektomija lahko koristno pri bolnikih z anemijo, levkopenijo, trombocitopenijo ali ko vranici povzroča nelagodje.
Niemann-Pickova bolezen
Bolezen Niemann - trapez - sfingolipidoz nezadostno aktivnost sfingomielinaze, kar povzroči kopičenje sfingomielina (ceramidi fosforilholina) retikuloendotelijskega celice povzročile.
Bolniki z tipa A so <5% нормальной активности сфингомиелиназы. Заболевание характеризуется гепатоспленомегалией, задержкой развития и быстро прогрессирующей нейродегенерацией.
Pri bolnikih z aktivnostjo sfingomielinaze tipa B je 5-10% normalne. Tip B ima različne klinične manifestacije, kot tipa A se lahko razvijejo limfadenopatija in hepatosplenomegalija. Pancitopenija precej pogosta. Večina bolnikov s tipom B imajo malo ali nič nevrološke okvare in preživeti v odrasli dobi, so lahko klinično ne razlikuje od bolnikov z Gaucherjevo boleznijo tipa I. V hujših primerih, kot na primer napredni pljučni infiltrati povzroči velike zaplete.
diagnostika
- Prenatalna pregled.
- Študija sfingomielinaze levkocitov.
Obe vrsti sta ponavadi ne zavedajo anamnezo in rezultatov raziskav na prvem mestu hepatosplenomegalija. Diagnozo lahko potrdimo z analizo levkociti sfingomielinaze, in to je mogoče storiti pred rojstvom skozi amniocenteza ali horionskega vzorčenja villus.
zdravljenje
Presaditvi celic kostnega mozga ali matičnih preiskovanih kot možnostmi zdravljenja.
Tay-Sachs bolezen in Sandhoffa
Tay - Sachs bolezni in Sandhoffa - sfingolipidozy s pomanjkanjem hexosaminidase povzroča, povzroči hude nevrološke simptome in zgodnjo smrt.
Gangliozidi so kompleksne sfingolipidi. Obstajata dve osnovni obliki, GM1 in GM2, ki so lahko vključeni v lizosomskih bolezni nakopleniya- razlikovati dve glavni vrsti GM2 gangliosidosis, od katerih jih je več različnih mutacij lahko povzroči.
Tay - Sachs bolezen. Dedovanje je avtosomno recesivno, najpogostejše mutacije odkrite v 1/27 normalno odraslih Evropske judovske (Aškenazi) rodu vzhoda, čeprav druge mutacije odkrili pri nekaterih populacijah francosko-kanadskega prebivalstva in Cajun.
Glavne faze razvoja otrok s Tay - Sachs so se začeli zaostajati po starosti 6 mesecev, hkrati pa razvija postopno poslabšanje kognitivnih in motoričnih funkcij, ki vodi do razvoja napadi, duševna zaostalost, paraliza in smrt pri otrocih, mlajših od 5 let. Češnjevo rdeča makleznaya izpuščaj je pogost simptom.
V odsotnosti učinkovitega zdravljenja se osredotoča na upravljanje reproduktivnih sposobnih odraslih, ki prikazujejo v ogroženih skupin za odkrivanje nosilcev (s testiranjem encimske aktivnosti in prisotnost mutacij) v kombinaciji z genetskega svetovanja.
Krabbe bolezen
Krabbe bolezen sfingolipidozom, ki povzročajo duševno zaostalost, paraliza, napreduje do smrti.
Krabbe bolezen (galactosylceramide lipidoze, leukodystrophy Globoid celic) povzroča pomanjkanjem avtosomno recesivna galactocerebroside galaktosidaze.
Ta bolezen prizadene otroke in je značilna duševna zaostalost, paraliza, pseudobulbar paralizo, postopno do smrti.
Ker je presaditev kostnega mozga učinkovito zakasni pojav simptomov, ki se včasih izvaja prenatalni diagnozi ali prebiranje novorojenčkov (rutinsko New York).
metachromatic leukodystrophy
Metachromatic leukodystrophy sfingolipidozom je povezana s pomanjkanjem arilsulfataza, ki povzroča progresivno paralizo in demence, ki vodi do smrti v starosti 10 let.
Ko metachromatic leukodystrophy (sulfatide lipidoze) pomanjkljivost arilsulfataza povzroča kopičenje lipidov v metachromatic bele snovi centralnega živčnega sistema, perifernega živci, pochkah- kopičijo v živčnem sistemu povzroči centralnega in perifernega demielinizacijo. Obstajajo številni mutatsii- pacienti ločeni glede na starost, začetku in hitrosti napredovanja bolezni.
Za zgodnji oblika je značilna progresivna paraliza, in demenca, običajno pojavi pred starostjo 4 let in vodijo do smrti v približno 5 let po pojavu simptomov. Mladoletnike oblika se prične pri starosti od 4 do 16 let z motnja hoje, intelektualnih motenj in simptomov periferno nevropatijo. V nasprotju z zgodnjim obliki globoke tetive refleksi so ponavadi zasedeni. Na voljo je tudi mehkejša oblika za odrasle.
Diagnozo je predlagal kliničnih simptomov in odkrivanje omejeno prevodne hitrosti živcev.
Učinkovito zdravljenje ne obstaja.
Fabryjeva bolezen
Fabry bolezen s pomanjkanjem galaktozidaze sfingolipidozom povzročil.
Fabry bolezni (angiokeratoma Corporis diffusum) je-X povezana pomanjkljivost lizosomskega encima galaktozidazo, ki je potreben za normalno trigeksoziltseramida razgradnjo. Glikolipid (globotria-oziltseramid) nabirajo v mnogih tkivih (npr endotelija krvnih žil, limfnih žil, srca, ledvic).
Diagnoza pri moških, ki temelji na pojavu tipičnih lezij (angiokeratomas) v spodnjem delu telesa in značilnostmi periferne nevropatije, pomotnenja roženice in ponavljajočih se vročinskih epizod. Smrt je posledica ledvične odpovedi ali srčne in cerebrovaskularnih zapletov. Heterozigotnim ženske so ponavadi brez simptomov, lahko pa imajo oslabljen obliko bolezni.
Zdravljenje - -galakgozidazoy encimsko nadomestno rekombinantni (beta galaktozidaza) v kombinaciji s podpornimi ukrepi v mrzlico in bolečine. Presaditev ledvice je učinkovita pri zdravljenju odpovedi ledvic.
holesterol bolezen skladiščenja estra in bolezni Woolman
Holesterol bolezen skladiščenja estra in Woolman sfingolipidozami bolezni so ga pomanjkanjem lizosomov kisla lipaza povzročil, zaradi hiperlipidemije in hepatomegaliji.
Te bolezni - redka avtosomna recesivna bolezni, ki izhajajo iz akumulacije estrov holesterola in trigliceridov, predvsem v lizosomih histiocytes, kar povzroči pojav penastih celic v jetrih, bezgavke in drugih tkiv.
Woolman bolezen - huda oblika, ki se kaže v prvem tednu življenja, slabega apetita, bruhanje, trebušne distention in sekundarni gepatosplenomegalii- dojenčki običajno umre v roku 6 mesecev.
kopičenje holesterola estrov bolezni je manj huda in se lahko kaže na več, tudi odraslost, če se lahko dokaže, gepatomegaliya- lahko razvijejo prezgodnje ateroskleroze, pogosto težka.
Diagnoza temelji na pomanjkljivosti klinično lipaze znaki kisline v vzorcih jetrni biopsiji ali kultiviranih kožnih fibroblastov, limfocitov in drugih tkiv. Pred rojstvom diagnoza temelji na odsotnosti kisla lipaza aktivnosti v kultiviranih horionski resic.
Ni učinkovitega zdravljenja, vendar statini zmanjšajo plazemske koncentracije LDL.
 Bolj razširjeni polip v želodcu
Bolj razširjeni polip v želodcu Spermiogistogenez. Spermatide in njihove značilnosti
Spermiogistogenez. Spermatide in njihove značilnosti Diagnoza sfingomielinoza. organi spremenijo NPD
Diagnoza sfingomielinoza. organi spremenijo NPD Mucolipidosis: mannozidoz in fucosidosis. Juvenilni sulfatidoz tip oustina
Mucolipidosis: mannozidoz in fucosidosis. Juvenilni sulfatidoz tip oustina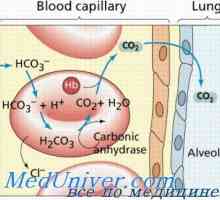 Amavroticheskaya prirojeno Idiotizam. Mucolipidosis
Amavroticheskaya prirojeno Idiotizam. Mucolipidosis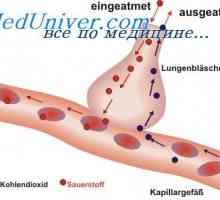 Diagnoza sulfatidoza. Sfingomielinoz Niemann-Pickova bolezen
Diagnoza sulfatidoza. Sfingomielinoz Niemann-Pickova bolezen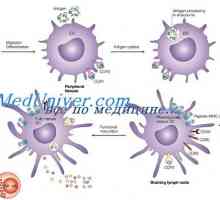 Učinek imunomodulator na dendritične celice. Morfologija dendritične celice
Učinek imunomodulator na dendritične celice. Morfologija dendritične celice Folikularni velikocelični rak adenokarcinom in alveolarni ščitnice
Folikularni velikocelični rak adenokarcinom in alveolarni ščitnice Zlatenica pri novorojenčkih z bakterijskimi okužbami, klinične manifestacije, diagnoza
Zlatenica pri novorojenčkih z bakterijskimi okužbami, klinične manifestacije, diagnoza Izvornih celic presajena za kopičenje bolezni in talasemije
Izvornih celic presajena za kopičenje bolezni in talasemije Bolezni krvotvornih sistema. Gaucherjeva bolezen
Bolezni krvotvornih sistema. Gaucherjeva bolezen- Gaucherjeva bolezen se nanaša na lipidov- akumulacije bolezni sfingolipidozam zaradi okvare gena,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Terapija, bolezni prebavnega sistema
- Prašiči celice se uporabljajo v umetnem jetrih
 Kršitev presnovo lipidov
Kršitev presnovo lipidov Gaucherjeva bolezen, simptomi, zdravljenje
Gaucherjeva bolezen, simptomi, zdravljenje Niemann-Pickova bolezen
Niemann-Pickova bolezen Kongenitalna fibroza jeter
Kongenitalna fibroza jeter Izmenjava prekrški kovine
Izmenjava prekrški kovine Metaboličnih motenj presnove sečne kisline in kovine
Metaboličnih motenj presnove sečne kisline in kovine