Mišična distrofija pri otrocih: simptomi, zdravljenje, diagnoza, vzroki

Vsebina
Mišične distrofije so podedovali progresivna mišična sistemske bolezni, ki nastanejo zaradi napak v enem ali več genih, ki so potrebni za normalno delovanje mišic.
Zanje je značilno selektivno razširjanje šibkosti in posebne narave genetskih nepravilnosti.
Becker distrofija, čeprav tesno povezana, je poznejši začetek in povzroča blažje simptome. Druge oblike vključujejo distrofija, Emery - Dreyfusa, miotonična distrofija, telo obroča distrofija, fatsioskapulogumeralnuyu distrofija in prirojenih distrofija.
Duchennova mišična distrofija in Becker mišična distrofija
Duchennova mišična distrofija in Becker mišična distrofija je-X vezana recesivna bolezen označena s progresivnim oslabelost proksimalnih mišic z degeneracijo mišičnih vlaken povzročajo. Becker distrofija je pozen začetek in povzroča blažje simptome. Zdravljenje je ohranjanje funkcije na fizično terapijo, naramnice in ortopedsko uporabo apparatov- prednizona je predpisana pri nekaterih bolnikih s hudo funkcionalno okvaro.
Ko se to mutacijo Duchennova mišična distrofija vodi v hudo pomanjkanje (<5%) дистрофина, белка мембраны мышечных клеток. При дистрофии Беккера мутация приводит к образованию ненормального дистрофина или малому его количеству. Дистрофия Дюшенна поражает 1/3000 родившихся мужчин. Дистрофию Беккера выявляют у 1/30 000 родившихся мужчин. У женщин-носителей может быть выражено бессимптомное повышение уровня креатинкиназы и, возможно, гипертрофия задней части голени.
Simptomi in znaki
Duchennova distrofija. Ta motnja se običajno pojavi v starosti od 2-3 let. Šibkost vpliva proksimalne mišice, kot pravilo, prvih spodnjih okončin. Otroci pogosto gredo na prste se waddling hoja in lordoza. Napredovanje šibkosti nenehno razvijali ud skrcenja kontraktur in skoliozo. Razvija zanesljivo psevdogipertrofiya. Večina otrok je priklenjena na invalidski voziček v starosti 12 let. Srčna prizadetost je običajno brez simptomov, čeprav ima 90% bolnikov, ki so motnje EKG. Ena tretjina od njih ima svetlo non-progresivno duševnem razvoju, ki vplivajo na verbalno sposobnost je močnejši od izvedbe.
Becker distrofija. Ta motnja je ponavadi kaže simptomatsko veliko kasneje in lažji. Sposobnost, da se premaknete se običajno hranijo najmanj 15 let, in veliko otrok se gibljejo v odrasli dobi. Večina žrtev živi do 30 in 40 let.
diagnostika
- Distrofinski imuno. I Analiza DNK za mutacije.
Diagnozo domnevala, odvisno od značilnih kliničnih simptomov, starosti ob nastopu in družinske zgodovine, ki vključujejo-X vezana recesivno dedovanje. Myopathic spremembe opazili na elektromiografijo (potenciali motorno enoto hitro narašča, so kratkotrajni in majhno amplitudo) in mišičnih biopsij (nekroze in označeno velikostjo spremembe mišičnih vlaken, niso ločeni od motorne enote). kreatin kinaze ravni presegel 100-krat normo.
Diagnoza je potrjena z analizo distrofinske z imunskim barvanjem biopsijo. Distrofija pri bolnikih z Duchennovo mišično distrofijo ne zazna. Analiza mutacij DNA v levkocitov periferne krvi lahko potrdi tudi diagnozo pri ugotavljanju kršitve v distrofinskem genu (delecijami ali podvajanjem ima približno 65% bolnikov, in točkovne mutacije - približno 25%).
zdravljenje
- Podporne ukrepe.
- Včasih prednizona.
- Včasih korektivnih kirurgija.
Posebno zdravljenje ne obstaja. Zmerna vadba je priporočljivo opraviti čim dlje. Gleženj preprečuje upogibanje med spanjem. Ortopedski pripomočki na stopalih lahko začasno pomaga ohranjati sposobnost stati in hoditi. Izogibajte ozhireniya- potrebe kalorični so verjetno nižje kot običajno. Prikaz genetsko svetovanje.
Dnevni vnos prednizolon bistveno ne podaljša klinično izboljšanje, vendar pa lahko upočasni potek bolezni. Soglasje o dolgoročni učinkovitosti ni tam. Genska terapija se še ni razvila. Popravljanje operacije so včasih potrebni. odpoved dihal včasih lahko zdravimo z uporabo neinvazivno podpore dihanju (preko nosno masko). Izbirni traheotomijo bolj znano, da omogoča otrokom z Duchennovo mišično distrofijo živi v starosti 20 let.
Druge oblike mišične distrofije
Distrofija Emery - Dreyfus. Ta bolezen se lahko deduje avtosomno dominantno, avtosomno recesiven (zelo redko) ali X vezan tipa. Skupna pogostnost ni znana. Ženske so lahko nosilci, ampak samo moški trpijo zaradi simptomatskega bolezen-X vezana dediščino. Geni povezani z distrofijo, Emery - Dreyfus, kodiranje jedrsko membranskih proteinov Lamina A / C (avtosomnem) in emerin (X-vezan).
Manifestacije mišične šibkosti in izčrpanosti se lahko pojavijo kadarkoli pred dopolnjenim 20 letom starosti in ponavadi vplivajo na biceps, triceps in manj mišic distalni noge. Srce pogosto povezane z atrijsko motnjami paraliza prevajanja (atrioventrikularni blok), kardiomiopatije in nenadna smrt veliko verjetnostjo.
Diagnoza kliničnih podatkov kaže, starost nastopa bolezni, in družinske zgodovine. Kot tudi rahlo povečanje serumskih ravni kreatin kinaze in myopathic funkcije na elektromiografijo in mišično biopsijo. Diagnozo je potrjeno s testi DNK.
Zdravljenje vključuje terapijo, katere cilj je preprečevanje kontraktur. Srčni so včasih ključnega pomena za bolnike z nenormalno prevajanja.
miotonična distrofija. Miotonična distrofija - najpogostejša oblika mišične distrofije med belci. To se zgodi s frekvenco približno 30/100 000 živorojenih otrok, moških in žensk. Dedovanje je avtosomno dominantna z različnimi penetrantnih. Dve genetska loci - DM 1 in DM 2 - povzroči anomalije. Simptomi in znaki začeli v adolescenci ali zgodnji odrasli dobi in vključujejo miotonija (sprostitev takoj po krčenje mišic) izčrpavanje in šibkost distalnih udov mišic (predvsem dlani) in obraza (še posebej razširjena ptoza) kardiomiopatijo. Prav tako se lahko razvije duševna zaostalost, sive mrene in endokrine motnje.
Na diagnozo kaže značilne klinične ugotovitve, starost ob nastopu in družino anamnez- potrjeno diagnozo testiranja DNK. Zdravljenje vključuje uporabo ortopedskega pripomočka ob vznožju in terapijo z drogami previs miotonija (npr meksiletin od 75-150 mg peroralno 2-3-krat dnevno).
Distrofija, ud pas. Trenutno znani podtipi izoliranega 21 obroča distrofije: 15 avtosomno recesivna in avtosomno dominantna 6. Skupna pogostnost ni znana. Več kromosomske loci so bile ugotovljene za avtosomalno dominantno (5q [genski produkt ni znana)) in recesiven (2q, 4q [[3-sarcoglycan] 13q [-sarkoglikan] 15q [kalpainskega, kalcijevega aktivira proteaze] in 17Q [-sarkoglikan ali adgalin]) oblike. Konstrukcijska (npr-distrofinski povezan glikoprotein) ali nestrukturiran (npr proteaze) lahko vpliva proteinov.
Simptomi vključujejo slabost v pasovih in proksimalnih delov okončin. Pojav giblje od zgodnjega otroštva do odrasle življenjsko začnejo augosomno recesivno tipa, ponavadi v otroštvu, in te vrste so v glavnem povezana s poškodbami medeničnega obroča.
Na diagnozo kaže značilne klinične ugotovitve, starost ob začetku in družina anamnez- diagnoza zahteva tudi določitev histologijo mišico imunocitokemija, Western blot analizo in genetskim testiranjem za prisotnost določenih proteinov.
Zdravljenje je namenjeno preprečevanju kontraktur.
Fatsioskapulogumeralnaya distrofija. Nastop v adolescenci ali mladi odrasli dobi je značilno počasno napredovanje: otrok težko žvižgati, da zaprejo oči in dvignejo roke (zaradi šibkosti mišic, ki stabilizirajo lopaticami). Pričakovana življenjska doba je normalno. Zgodnji sprememba, značilna po šibkosti obraz, ramena in nosilni pas, hitro napreduje.
Na diagnozo kaže značilne klinične ugotovitve, starost ob nastopu in družino anamnez- potrjeno diagnozo analize DNK.
Zdravljenje sestoji fizikalne terapije.
Kongenitalna mišična distrofija. To ni samostojna motnja je detektirati prirojena motnja, povezana z eno izmed več redkih oblik mišične distrofije. Diagnozo sumimo kateremkoli novorojenčka počasen, vendar je treba ločiti od prirojene miopatije uporablja mišično biopsijo.
Zdravljenje je sestavljen fizikalne terapije, ki lahko pomagajo ohraniti delovanje mišic od.
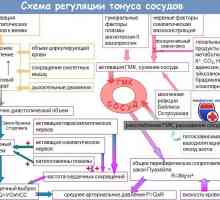 Prirojena miotonija Thomsen bolezen. Miotonična distrofija Krabbe sindrom
Prirojena miotonija Thomsen bolezen. Miotonična distrofija Krabbe sindrom Progresivna mišična distrofija. Oblike in simptomi mišične distrofije pri otrocih
Progresivna mišična distrofija. Oblike in simptomi mišične distrofije pri otrocih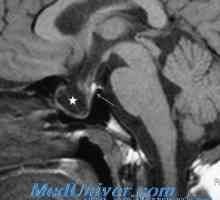 Diagnostiko in diferencialno diagnozo adipozo-genitalni sindrom
Diagnostiko in diferencialno diagnozo adipozo-genitalni sindrom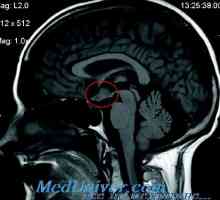 Adipozo-genitalnih distrofija: spremembe morfologija organov
Adipozo-genitalnih distrofija: spremembe morfologija organov Refleksna simpatična distrofija pri otrocih. Rodonalgia in ponavljajoče polihondritis
Refleksna simpatična distrofija pri otrocih. Rodonalgia in ponavljajoče polihondritis Primarni distrofija roženice
Primarni distrofija roženice- Sekundarni distrofija roženice
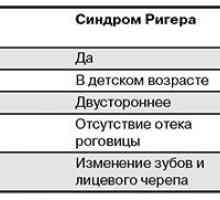 Rieger sindrom. Peters anomalija
Rieger sindrom. Peters anomalija Timing prenatalni diagnozo genetske bolezni.
Timing prenatalni diagnozo genetske bolezni. Diagnozo izoliranih blastomeres. Diagnostična patologija na stopnji blastociste.
Diagnozo izoliranih blastomeres. Diagnostična patologija na stopnji blastociste.- Postopno mišična distrofija-Bistvena progresivna degeneracija mišičnega tkiva, izhaja iz katerega…
- Kalijev orotat (kalii orotas). Kalijeva sol uracil-4-karboksilne (orotatna kislina). Besede:…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Genetske bolezni so nekatere izmed najpogostejših bolezni
- Enostavno - in roženice presaditev ne potrebujete
- Pametni telefon spremlja učinkovitost zdravljenja z mišično distrofijo
- Viagra pomaga z Duchennovo mišično distrofijo?
 Adipozo spolnih distrofija
Adipozo spolnih distrofija Akutna distrofija jeter
Akutna distrofija jeter Pigmentnim degeneracija mrežnice zdravljenja oči
Pigmentnim degeneracija mrežnice zdravljenja oči Miotonična distrofija
Miotonična distrofija