Prirojene okvare v noč in perifernega vida

Vsebina
mirovanju motnja
Prirojena mirovanju nočno slepoto
Ko je patologija opazili neprogresivne nočno slepoto (dnevno slepoto) in temno prilagajanje, ki ga povzroča droga-System funkcionalne motnje je genetsko heterogena bolezen. Morda dedovanje avtosomno dominantno ali recesivno tipa, in dedovanje-vezan na kromosom X. Razvoj bolezni je povezana z mutacijami genov, ki kodirajo sintezo procesov, prevoz in skladiščenje rhodopsin.
Več ločenih oblik bolezni.
1. Prirojene mirovanju nočno slepoto z normalnim oči spodaj:
- Tip Riggs (tipa I);
- tip Schubertov-Bornstein (tipa II) - Obstajata dva podtipa: popolna disfunkcija strani batnice sistema in preostalo funkcijo.
2. Prirojena miruje noč slepota s spremembami Fundus:
- Ogushi bolezen - I in tipa II;
- belotochechnoe eyeground;
- Opazila mrežnica iskrenost.
Očesni simptomi. Zmanjšana amplitudo obeh valov. Ritmična ERG pri 30 Hz frekvenco se ne spremeni. Krivulja temno prilagoditvenem enofazna brez kakršnih koli stožec paličasto-oblikovane zavoja. Ostrina vida se navadno ne spremeni, le redko nekoliko zmanjšala. Značilnosti rhodopsin po reflektometrijo normalno. Bolezen je dedna v recesivna spol vezana tipa.
Kongenitalna stacionarno nočno slepoto normalno oko tipa spodnji II (Schubert-Bornstein) je značilno pomanjkanje ali močan padec b valovnega regulatorjev obe vrsti pri normalnem ali povečane amplitude vala ( "negativno negativno" ERG).
S polno vrste prirojeno stacionarno nočno slepoto Schubert-Bornstein scotopic ERG odsotnega Fotopični - shranili ali subnormalni. Največja ERG minus negativen. Dark prilagoditev močno oslabljena. V kombinaciji s srednje in visoko kratkovidnost obsegu slabovidnosti in nistagmus.
Nepopolna vrste prirojeno stacionarno nočno slepoto Schubert-Bornstein je povečanje temno prilagoditev pragovi 1.0-1.5 dnevnik. U Scotopic ERG zniža za maksimalno ERG označen rahlo nižje v amplitude vala in izrazito zmanjšanje b-val, tj značaj shranjenih negativen negativna regulatorjev. Z nekaj kovinski refleksno oftalmoskopom mogoče opaziti v osrednjem delu mrežnice in obrobjem. Ostrina vida se lahko bistveno zmanjša. Refrakcija lahko tako emmetropic in ammetropicheskoy.
Tip Schubert-Bornstein je dedna v recesivna spol vezana tipa.
Video: Pogled center "mikrokirurgija". mrežnice patologija očesa
Prirojena miruje noč slepota s spremembami Fundus
Ogushi bolezni (Oguta, Oguchi) - Dvostransko dedna bolezen, ki temelji na obsežnih degenerativnih spremembah palice in stožčasto, brez vključevanja v patološkem postopku retinalnih pigmentnega epitelija. Dedovanje - autosomnoretsessivnoe. Prizadene predvsem japonski spust.
Vzrok bolezni velja, da preneha z razvojem fotoreceptor oči enote na ravni običajne 7-8 meseca starega zarodka.
Manifestih bolezni v zgodnjem otroštvu je skoraj popolna slepota ponoči, zmanjšano prilagoditev oči v mraku in barvno motnjami vida. Oftalmoskopom opozoriti pepela sivo ocesno ozadje s kovinskim sijajem, temno senco plovila na fundusa obodu razmeroma temne makularni območje rumenkaste lise na očesnem dne- pozitivnim simptomom Midzuo (obnavljanje normalnega barvnega ocesno ozadje, potem ko je v temnem prostoru). Te spremembe se pojavljajo predvsem na območju ekvatorja. Ostrina vida ostane normalno ali rahlo zniža, periferni vid v normalnih mejah. Zmanjšanje scotopic in največ (zaradi komponenti paličastega) ERG Fotopični ERG normalno. pigmentne lastnosti niso spremenile.
Obstaja več vrst bolezni. Pri tipu I dvofazna temno prilagoditev krivulje opazili med podaljšanim bivanjem v temi (2-3 h do 24 h), s tipom II temno prilagajanje krivulje je enofazna narave.
Histološko razkrila precej degeneracijo palic in stožcev.
Belotochechnoe fundus značilna po paramakulyarno videz in srednje periferni mrežnici multiplo belega žarišč v obliki točkovnih postavljeno mestih, ki se nahajajo v globlje plasti mrežnice. Pri PAH razkrivajo več manjših fenestrated pomanjkljivosti pigmentnega epitelija v srednjem obodu mrežnice, toda za razliko od Druse giperflyuorestsentsii niso opazili te pomanjkljivosti. Na dolgo ostali v temi, temno pragov za prilagajanje lahko doseže normalno raven, čeprav so pri nekaterih bolnikih, tudi v tem primeru, so ostala povišana. Scotopic ERG lahko normalno ali subnormalni negativen negativen. Med podaljšanim temno prilagajanje ERG amplituda je pravilna. Značilno pojemka regeneracija vizualne pigmenta. To je lahko povezano z Alportovim sindromom.
Histološka preiskava je pokazala kopičenje fustsina zrnc v celicah pigmentnega epitelija in atrofijo njegovih odsekov. Način dedovanja - avtosomno recesivno.
Opazila mrežnica Candor značilen pojav v globokih plasti mrežnice multiple žarišč nepravilne oblike z jasnimi mejami umazano rumeno. Spremembe so lokalizirana predvsem v območju ekvatorja. Ko PAH pojavlja v žarišč giperflyuorestsentsiya. Fotopični ERG se ni spremenilo. Amplituda scotopic ERG zmanjša. Vendar pa je po dolgem bivanje v temi, je amplituda scotopic ERG in krivulja temno prilagajanja doseči standarde. Tako kot v fundusa belotochechnom kinetike reduciranega vizualne pigmenti. Ostrina vida in barvni vid niso spremenile. Dedovanje pojavi v avtosomno recesivna način.
Postopno chorioretinal distrofija
Tapetoretinalnoy mrežnice abiotrophy (sin. Retinitis pigmentosa)
Morfološko odkrivanje uničenje palic in stožcev s svojim zunanjim segmentov vacuolization, izginotje jedrskih in plexiform plasti pigmentnih celic migrirajo v notranje plasti mrežnice, širjenje glialnih celic in vlaken, ki zapolnjujejo izpraznjenega prostora, fibroze in glioze žilnice in mrežnice plovila, horoidna atrofija.
Več ločenih oblik bolezni.
1. Genetski vrste:
- avtosomno dominantna vzorec 9-43% vseh primerov pigmentnim degeneracije mrežnice. Značilno je pozno pojav simptomov, počasno napredovanje;
- Avtonomno recesivna - najpogostejšo obliko bolezni (20-35% vseh primerov). Značilno je zgodnji začetek;
- X-vezana vrsta - je najhujša oblika retinitis pigmentosa, razvija pri moških in vodi v popolno izgubo vida v zadnjem desetletju. Ženska, nosilec bolezni pogosto znake rahlih mrežnice poškodb. Ta oblika je na voljo v 8-45% primerov;
- občasna tip se nahaja v 23-48% primerov.
2. Anatomski vrste:
- Tipičen - značilne razpršene narave sprememb (hipo-pigmentacije in pigment migracije začne vsredneekvato-ble cono, postopoma širi od centra). Spremembe v rumeni pegi, majhen, čeprav so v kasnejših fazah lahko povzročijo degeneracijo "središče tarče";
- netipično - lahko nadaljuje na več načinov:
- razpršene spremembe se lahko v kombinaciji z zgodnjim bolezenskega procesa, ki vključuje makule območje;
- sektorske spremembe - kopičenje pigmenta v obliki polmeseca na dnu časovnega območja;
- Enostranska retinitis pigmentosa - Dvostransko asimetrična značilnost lezije pozno v patološkega procesa, ki vključuje kolegi oko.
3. Funkcionalne motnje:
- Enostavno postopno tip - tip dedovanja je ponavadi prevladujoč. Značilnost počasnega napredovanja ohraniti vidno ostrino do 60 let;
- zmerni z napredovanjem v višji stopnji - označen s recesiven dedovanje. Za 60 let razvija vizijo cevi in poslabšanje vida;
- grobo napredovanje - značilnost-X vezan način dediščine. Izguba funkcije vida obstaja že 40 let.
Očesni simptomi. Zmanjšana temno prilagajanje. Obstaja značilni enotna obročasti zožitve meje periferno vidno polje do tvorbo cevastega. Poleg tega lahko pride do obročasti skotom, centralne, paracentral scotomas ali sektorska (z boleznijo centralnega ali sektorski oblikami) - obstaja znatno zmanjšanje ali odsotnost regulatorjev.
Za bolezen označena z odlaganjem pigment v obliki "kosti organov" in zmanjšanje količine zoženja mrežnice krvnih žil, voskasta bledo optični disk. Na začetku bolezni na periferiji mrežnice od ekvatorja obstajajo področja hipo- in razbarvanje, pa tudi sub in intraretinal belkasto-siva triki in parcel z rdečkasto odtenkom. Potem je točka in zrnate vključki ( "sol in poper"), nato stvori Značilna oblika kostnih celic. Postopoma so spremembe namnožijo iz obrobja v središče. Postopek je običajno dvosmerni simetrični a.
} {Modul direkt4
Spremembe v makule regiji se pojavljajo pri 63-74% bolnikov. Najpogosteje se pojavijo: stushevannost in izginotje fovealnega refleksa, epiretinal membrane atrofije in hipopigmentacija (v kasnejših stopnjah), pri čemer zrnatost ali madeži pigmentni epitel, vsaj - Tip distrofija "biki oči", makularni edem, cistična degeneracija in nekatere druge spremembe.
Označen s kombinacijo z miopija, katarakte napredovanje mrežnice sprememb.
prirojena amavroze prirojenih Leberjeva (sin. Tapetoretinalnoy otroci Leberjeva prirojene slepote)
Dedna bolezen, ki se kaže dvostransko retinitis retinitom- trenutno obravnavajo kot disnukleotsidoz z uničevanjem palice in stožci. Način dedovanja - avtosomno recesivna, čeprav je v nekaterih primerih, označenih s dominantnem dedovanju.
Klinični znaki in simptomi. Očesna spremembe so v kombinaciji z duševno zaostalost, izguba, različne nevrološke motnje, vključno z napadi sluha.
Histološko razkrivajo izoliranega uničenje pigmentnega epitelija in odsotnosti fotoreceptorjem plasti, namesto katerega je plast kockasta celic.
V tipični potek bolezni je zelo nizka vizija od rojstva. Otroci so plavajoče gibanje oči sunkoviti gibi oči, učencev inertno, brez pritrdilnih elementov. V zgodnjem otroštvu tam dan slepoto. Ponavadi v prvem letu življenja, sprememb v fundusa niso na voljo, v starejših letih ob robu dna oko zazna pigmentne depozitov v obliki "kostnih celic." V poznejših fazah bolezni pojavijo na mrežnica razpršena pigment "prahu" v običajni optični disk ali pigmenta displazijo degeneracije makule ali mrežnice pigment fenomen pigmentnih "kosti organov", redčenje in bledica plovila ONH. Refrakcija najbolj hyperopic z psevdootekom optičnega diska, vsaj - kratkovidna. Značilna odsotnost ERG. Opazovano astigmatizem, izmenično konvergentno škiljenje. Je globoko položaj v očesni votlini zaradi atrofijo mrežnice, retrobulbarnim.
Laurence-Luna sindrom, Bardet-Biedl (sin. Dientsefaloretinalnaya degeneracija)
Redka dedna bolezen, ki temelji na sekundarnem poškodbe mrežnice neuroepithelial abiotrophy in hipotalamus-hipofiza sistema. Način dedovanja - avtosomno recesivno.
Klinični znaki in simptomi. Bolezen se kaže v prvem letu življenja postopoma debelih gipogenitalizme, duševna zaostalost, polydactyly. Možne podvozja deformacije, syndactyly, ravno, gluhost in gluho-turricephaly, pritlikavost ali gigantizem, zobne motenj, bolezni srca in ledvična displazija hipoplazija, nefroskleroza, hidronefroz, pielonefritis, analni atrezija. Pri moških obstajajo impotenca in azoospermiji ginekomastije, kriptorhizm- ženske - vaginalni atrezija, hipoplastična jajčnikih, oligo-in amenoreja. Kot rezultat hormonskih motenj ravni metabolnih procesih se zmanjša. Včasih je nevrološka motnja. Pričakovana življenjska doba ne presega 40 let.
Morfološko identifikacijo možganske okvare - atrofija ovoje agenesis v corpus callosum, asimetrije na polobli, hidrocefalusa. Hipotalamus jedra kažejo degeneracija z zmanjšanjem števila ganglijskih celic in rast vezivnega tkiva.
Očesni simptomi. Značilna razvoj dvostranskega pigmentnim degeneracija mrežnice, ki se kaže v različnih kliničnih oblik, kot tudi nočno slepoto in znižanem videnje različni meri. Označene degenerativne spremembe v makule regiji refrakcijsko napako, strabizem (navadno konvergira), mikroftalmijo, katarakte, atrofija vidnega živca. Spremembe v fundusa določena z visokim variabilnosti: majhna pigmentiran okrogla žarišča ali nepravilne oblike, raztresene okoli dna z očmi (značilno obliko bolezni) - tipično pigmentnim degeneracija z odlaganjem pigment v obliki "kostnih organov" v obodu optike DNA-manj pigmentnoznega degeneracijo brez pigmenta- retinalne degeneracije včasih zgodi v več žarišč brez belega pigmenta, razpršene po spodnjem očesa, - degeneratio retinae punctata albescens.
Diferencialna diagnoza preživeti s sindromi Alstrema-Halgrena, Graefe, Usher, Prader-Willi, akrotsefalopolisin-daktilska.
Horioderemiya
Nanaša se na dedne bolezni, v katerem je hkratno poraz mrežnice in žilnice. Tipičen-X vezano dedovanje. Bolezen se pojavlja v polovici sinov žensk - nosilci gena. Ženske nosilci gena sprememb v Fundus blagi, ostrina vida se ni spremenilo. Histološka preiskava pokazala sprememb ugodne pigmentni epitelij in fotoreceptor. Žilnice na področju pomanjkanja pigmentnega epitelija spremenile.
Očesni simptomi. Značilen pojav v motenj v zgodnjem otroštvu in temno prilagoditev na področju sprememb prikaza. Ostrina vida, se je bistveno zmanjšalo na 30 let. Na začetku bolezni obstaja več skotom in slepa širitev mesto. Postopno koncentrična zožitev vidnega polja vodi do tvorbe cevi. Barvni vid je normalno ali pa acyanopsia ali tritanomaliya. ERG ni registriran ali ima mikroERG.
Rahlo zoženje arterij, se žile niso spremenile. Četrtina bolnikov prisotne bledica optičnega diska.
Atrofije girate (sin. Lobular atrofija)
Nanaša se bolezni z generalizirano atrofijo mrežnice in žilnice. Razvoj bolezni s pomanjkanjem encima ornitinaminotransferazy povzročil. Način dedovanja - avtosomno recesivno. Obstajata dve genetske oblike: občutljivih in neobčutljivih vitaminu B6. Bolezen izvira iz mutacij na kromosomu X.
Klinični znaki in simptomi. V tekočine krvi in druga telesa povečano stopnjo ornitina. Krvna plazma je zmanjšal nivo glutamat, glutamin, kreatinina in Licinius. Ob proksimalno degeneracija skeletnih mišic, spremembe v EEG in EKG. Značilnost je prisotnost bolnikom z redkim ravne lase.
Očesni simptomi. Spremembe v fundusa pojavi v prvem desetletju življenja in se postopoma širijo iz obrobja v središče. Značilen pojav žarišč atrofije z zaobljenih robov. V zgodnjem otroštvu je nočna slepota in naglušne periferni vid. EWG ni registriran.
Opazila mrežnice abiotrophy
Alportovega sindroma (sin. GR-oculo-ledvičnim sindromom)
Dedna bolezen, ki je odvisna od prevladujočega gena, lokalizirana na kromosom X. Bistvo sindroma je progresivna ledvična odpoved v kombinaciji z naglušnosti.
Z obliko dednega in klinične simptome še 6 vrste sindrom: avtosomna dominantna s klasično juvenilni tipa gluhotoy--X vezana recesivna gluhotoy- s-X vezana recesivna odraslih gluhotoy- sindrom, X vezana recesivna gluhosti brez odraslih sindrom in druga defektov- avtosomnem -dominantny sindrom gluhosti in avtosomnem trombotsitopeniey- retsissivny mladostniškega sindrom naglušnosti.
Klinični znaki in simptomi. Bolezen se lahko manifestira v zgodnjem otroštvu izgubo senzorinevralna sluha, razvojne okvare ledvic in sečevodov s hematurija, proteinurijo, bakteriurije. Odpoved ledvic pojavlja pogosteje pri moških - fantje navadno umrejo v najstniških letih.
očesni simptomi pojavijo pri 1/6 pacientov in očitne anomalije telo sprednjim ali zadnjim lentikonus- anteriorni ali posteriorni subkapsularno katarakta- simptom Krukenberga- degenerativne spremembe mrežnice v obliki številnih belkasto tochek- sferofakiya, mikrofakiya.
Video: Otvoritev zaščitnega urada za otroke
Biett distrofija (sin. Z mrežnice abiotrophy robno distrofija roženice)
Kompleks očesni simptomi distrofična. Dedna bolezen, redka, počasi napredujoča, medtem ko je poraz v obe očesi. Etiopatogenezi ni znan. Trpijo, moški in ženske vseh starosti.
Očesni simptomi. Bolezen se razvije v 3-4 desetletju življenja, kaže v obliki roženice lezija mejnega degeneracije. Roženice STROMA na limbusa pojavijo svetleče, rumenkaste igle in valovite lise. Potem distrofična Postopek razvija v žilnice in mrežnice, kjer sta Sklerotičan lezije tvorjen v obliki okroglih vložki ali "kostnih organov", ki dajejo oko spodaj rjavo. mrežnične vene zožitev manj izrazita kot pri drugih oblikah degeneracije. ONH dekolorirovan. Funkcionalne motnje, kot je definirano napak v vidnem polju, nočno slepoto, progresivne zožitve vidnega polja pred tvorbo cevi in znatnega zmanjšanja ostrine vida.
Diferencialna diagnoza preživeti z retinitis pigmentosa.
Drusen steklasta žilnice plošča
Redka dedna bolezen, frekvenca, pri kateri je 0,35 na 1000 bolnikih z očesno patologijo. Značilna motnjo normalne presnove v vmesnem mrežnice snovi in pigment epitelija celic samih. Način dedovanja - avtosomno dominantna s spremenljivo penetrantnih in ekspresivnosti.
patogeneza. Kot rezultat metaboličnih motenj v mrežnici, so zbrani v citoplazmi celic pigmentnega epitelija večinoma hialina netopne beljakovinskih snovi. Tam proteinoze posamezne celice ali skupine celic epitela. Grudice večinoma hialina snov združita, njihova reja obložene pigmenta epitelnih celic sosednjih zdravih cone. epitelija celice proizvod pigmentni - glamelinovye telo - a Druzi stekleno ploščo. Ti ležijo na steklasto ploščo (Bruch je membranski), ki je močno razmejen od njih. V velikih drusen celice imajo vretenaste oblike ali ravno obliko z občasnim jedra in majhno količino pigmenta zrn. Vsebuje netopnega kalcijevo sol. Kasneje se razvije osifikacije prijatelje.
Očesni simptomi. Ophthalmoscopically prikazani drusen kot rumenkast ali belkasto-rumenkasta žarišča z zaobljenimi ostrimi robovi, velikosti običajno ni večja od dvakratne premera vene v ONH. Druzi pogosto nahajajo v časovno strani optičnega diska in okoli makule območju. Nahajajo se na notranji površini membrane Bruch je v obliki majhnih žarišč. Včasih rastejo velika. V teh primerih je okoli vidnega pigmenta obroča, ki sestoji iz bolj razširjeni RPE celice.
Video: 2015/11/08 - Program "Šestintrideset in šest" (36.6)
Koloidna degeneracija mrežnice CANDOR
Distrofična bolezni mrežnice, ki prizadene ženske v starosti 30-40 let. Osnovnimi boleznimi so primarna degeneracija membrane in degeneracijo pigmentnega epitelija celic Bruch je.
Očesni simptomi. Bolezen se začne z zmanjšanjem temno prilagajanja. Kasneje, ko bolezen napreduje v različni meri zmanjša perifernega in centralnega vida. Ophthalmoscopically v paramakulyarnoy regiji kažejo zelo velike (do 1 / W DD) drusen ravno nepravilne podolgovate oblike. S premikom središče udarcev cone makule drusen napredovanje v procesu za več let.
Sjögrenov sindrom-Larsson
Dedne bolezni degenerativnega narave, označen s triado simptomov: prirojene ihtioze, duševne zaostalosti in spastični udov tetraparesis. Patogeneza ni jasno. Nekateri raziskovalci so povezani z razvojem bolezni s povečanjem nekaterih bolnikih urinskega izločanja kinurenina in histidin. Način dedovanja - avtosomno recesivno.
med napredujočo boleznijo, stabilna včasih. Predvidena bolj neugoden.
Očesni simptomi. Patološke spremembe lahko pojavijo oči ektropium, tanjšanje roženice, nastanek erozije in razjede roženice z morebitno perforacijo. Majhen delež bolnikov s sindromom kaže nekakšno pigmenta degeneracije z majhno belo žarišč v osrednjem območju.
 Hypophosphatasia plod. Orogenic displazija
Hypophosphatasia plod. Orogenic displazija Akromezomelicheskaya displazija. eykardi sindrom
Akromezomelicheskaya displazija. eykardi sindrom Osteogenesis imperfecta. Diagnoza in Prognoza osteogenesis imperfecta pri plodu
Osteogenesis imperfecta. Diagnoza in Prognoza osteogenesis imperfecta pri plodu Prevladujoči in recesivni alelov kromosomov. Avtosomalne dominante dedovanje
Prevladujoči in recesivni alelov kromosomov. Avtosomalne dominante dedovanje Avtosomno recesivno dedovanje. -X vezano dedovanje
Avtosomno recesivno dedovanje. -X vezano dedovanje Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji
Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji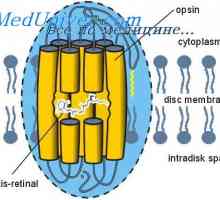 Svetlo in temno prilagoditev. Mehanizmi svetlimi in temnimi prilagajanje
Svetlo in temno prilagoditev. Mehanizmi svetlimi in temnimi prilagajanje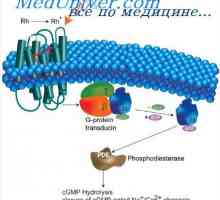 Slepota za posamezne barve. Funkcija mrežnice nevronov
Slepota za posamezne barve. Funkcija mrežnice nevronov Nekatere funkcionalne metode raziskovanja
Nekatere funkcionalne metode raziskovanja Kseroftalmije
Kseroftalmije- Degenerativnih bolezni mrežnice
- Pomanjkanje vitamina A (kseroftalmije, pomanjkanje retinol) nastane pri pomanjkanju vitamina A in…
- Dan-slepota (nočna slepota, nočna slepota) -rasstroystvo twilight vid. Etiologija prirojeno nočno…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
 Ključ eye2tv optimizira sliko za ljudi z barvno slepoto
Ključ eye2tv optimizira sliko za ljudi z barvno slepoto Kršitve presnove aminokislin
Kršitve presnove aminokislin Nevoid amentia
Nevoid amentia Albinizem pri ljudeh, oči
Albinizem pri ljudeh, oči Prirojene makularni spremembe
Prirojene makularni spremembe Monogenskih sindromov mvpr
Monogenskih sindromov mvpr