Mucolipidosis: mannozidoz in fucosidosis. Juvenilni sulfatidoz tip oustina
razen gangliosidosis GM1, Nadaljnje študije bodo poudariti tri nozokomialne oblike Mucolipidosis s sedežem encimski defekt - mannozidoz, fucosidosis sulfatidoz in juvenilnih (tip Oustina).
Mannozidoz - encim napaka je redek aktivnost a-manosidazna, ki posega v katabolizem manoze bogate odsek glikoprotsidov in celic (predvsem jeter in možganov) nabira oligosaharid bogata z manoze. Bolezen je opisan pri otrocih 2-4,5 let, kaže psihomotorične zaostajanje razvoja, hepatosplenomegalija, kyphoscoliosis, zamotnitev leče, vacuolization v limfocitih periferne krvi. Smrt nastopi v predšolskih letih.
Obdukcija študija nevronskih določene spremembe podobne spremembam v amavroticheskoy idiotizma lokalizirane na trupu in v možganski skorji, hrbtenjače, s pojavom demielinizacijo in glioze sledil. Spremembe v očesni mrežnici so odsotni. kopičenje celic najdemo poleg tega v jetrih, vranici, kostnem mozgu.
odloži snov delno topni v vodnih raztopinah, PAS pozitivne daje odziv lipidov. Elektronskim mikroskopska preiskava limfocitov in v nevronih odkrije povečane sferične lizosome - celično kopičenje.

fucosidosis - encim napaka je popolnoma inaktiviramo a-fucosidase in arilsulfataza A [Troost J. et al, 1977.] v možganih, jetrih in drugih organov, ki vodijo do kopičenja glikolipidov (tseramidoligosaharidov) vsebujejo fukoze sočasno akumulira in kisle mukopolisaharidi (hexuronic kislina). Dedovanje je avtosomno recesivno.
bolezen opisano v SIBS 2-3 let, označen s povečanjem spastične paralize, duševne zaostalosti, kaheksije.
ob fucosidosis opazovana kardiomegalija, atrofične spremembe kože in majhne spremembe hondrodistroficheskogo vrsto. Najdeno v urinu povečana fucoside vsebine v limfocitih periferne krvi - vacuolization. Obstajajo poročila dveh kliničnih vrst fucosidosis odvisno od stopnje aktivnosti encima [Troost J. et al., 1977].
spremembe možgani spomni sudanofilnuyu leukodystrophy s sočasno izgubo nevronov v možganih in male možgane. Lipidnos snov kopiči okoli krvnih žil. kopičenje celic najdemo poleg tega v jetrih, miokarda, ledvice, pljuča, koža, veznici očesa. Electron mikroskopske spremembe v kopičenje celic spominja Hurler sindrom (veliki vakuoliziranega Lizosomi) in lipidoze (prisotnost in koncentrične lamelne strukture). Histochemically vključkih oligosaharidov verige.
sposobni zaznati Pomanjkljivost a-L-fucosidase v kulturi dermalnih fibroblastov v celicah amnion in plodovnice, zaradi česar je mogoče nastaviti prenatalna diagnostika fucosidosis.
mladoletnike sulfatidoz (Tip Oustina) je kombinacija sprememb značilna metachromatic leukodystrophy (sulfatidoza) in gargoilizma (mukopolisaharidoza). pomanjkljivost bolezni harakteruzetsya delovanja encima arilsulfataza A, B in C v nasprotju s klasično metachromatic leukodystrophy, ki moti aktivnost samo arilsulfataza A pri otrocih, razen tistih, specifična za metachromatic leukodystrophy, pride do sprememb v obraznih kosti, prsi in dolgih kosti, tipično za Hurler sindroma. Urin dodeljena dermatan sulfata, heparan sulfat in sulfatides. Te snovi se odlagajo v tkivih.
Poleg teh oblik Mucolipidosis z določenimi encimski defekt opisane izvedbe tezaurismozov mešanega tipa, ki se med seboj razlikujejo kliničnih in morfološke značilnosti privede do različnih stopenj poškodbe skeleta in drobovja in različne stopnje poškodb osrednjega živčevja. Autosomes podedovala recesiven. Podrobnejši tri možnosti: tipa I, tipa II, tip III (psevdopolidistrofiya).
 Poslezheltushnaya encefalopatija. Microspherocytosis dedna bolezen Minkowski-Chauffard
Poslezheltushnaya encefalopatija. Microspherocytosis dedna bolezen Minkowski-Chauffard Agammaglobulinemia, X-vezana kromosoma, Bruton sindrom. Morfologija Bruton sindrom
Agammaglobulinemia, X-vezana kromosoma, Bruton sindrom. Morfologija Bruton sindrom Higashi - Chediak sindrom. sindrom Služba je pri otrocih
Higashi - Chediak sindrom. sindrom Služba je pri otrocih Glyukotserebrozidoz Gaucherjeva bolezen. Glikosfingolipidoz Fabry bolezen
Glyukotserebrozidoz Gaucherjeva bolezen. Glikosfingolipidoz Fabry bolezen Gangliosidosis. Sandhofa bolezni in mladoletnike gangliosidosis
Gangliosidosis. Sandhofa bolezni in mladoletnike gangliosidosis Diagnoza sfingomielinoza. organi spremenijo NPD
Diagnoza sfingomielinoza. organi spremenijo NPD Galaktoziltseramidoz Krabbe bolezen. Sulfatidoz metachromatic leykodistrosriya Scholz
Galaktoziltseramidoz Krabbe bolezen. Sulfatidoz metachromatic leykodistrosriya Scholz Amavroticheskaya prirojeno Idiotizam. Mucolipidosis
Amavroticheskaya prirojeno Idiotizam. Mucolipidosis Uolmana generalizirana xanthelasmatosis bolezni. refzuma sindrom
Uolmana generalizirana xanthelasmatosis bolezni. refzuma sindrom In leukodystrophies mukopolisaharidoze. Tveganje mukopolisaharidoze
In leukodystrophies mukopolisaharidoze. Tveganje mukopolisaharidoze Razvrstitev mukopolisaharidoze. Hurler bolezen, gentera, Sanfilippo, Morquio
Razvrstitev mukopolisaharidoze. Hurler bolezen, gentera, Sanfilippo, Morquio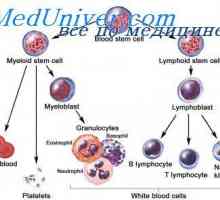 Izvor matičnih celic. embrionalne matične celice
Izvor matičnih celic. embrionalne matične celice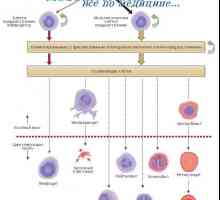 Vrste belih krvnih celic. Izvor belih krvničk
Vrste belih krvnih celic. Izvor belih krvničk Gaucherjeva bolezen se nanaša na lipidov- akumulacije bolezni sfingolipidozam zaradi okvare gena,…
Gaucherjeva bolezen se nanaša na lipidov- akumulacije bolezni sfingolipidozam zaradi okvare gena,…- Onkologiya-
 Kršitev presnovo lipidov
Kršitev presnovo lipidov Gaucherjeva bolezen, simptomi, zdravljenje
Gaucherjeva bolezen, simptomi, zdravljenje Niemann-Pickova bolezen
Niemann-Pickova bolezen Osteomieloskleroz
Osteomieloskleroz Neyrometabolicheskie bolezen
Neyrometabolicheskie bolezen Mucolipidosis
Mucolipidosis