Razvrstitev mukopolisaharidoze. Hurler bolezen, gentera, Sanfilippo, Morquio
Mukopolisaharidozo tipa I (Hurler bolezen) -nasledovanie avtosomno recesiven. Celice in medceličnem snov poseda dermatapsulfat, heparan sulfat, in gangliozidi. Obstaja pomanjkanje encima beta-galaktozidaze, in poslednimdannym in-iduronidazy.
Tam so bile hude simptome gargoilizma s progresivnimi duševnimi motnjami, zgodnje motnjo roženice, gluhosti in Bolezni jeter, Splenomegalija. V krvnih limfocitov z granule z metachromatic v urinu močno poveča vsebnost mukopolisaharidov. Otroci umirajo v starosti 10-12 let, s simptomi možganske poškodbe (hidrocefalus), srca ali iz sočasnimi okužbami.
Mykopolisaharidoz tipa II (Gentera bolezen) -nasledovanie recesivna, povezan z X-hromosomon, deponiran v tkivih dermatan sulfata in heparan sulfat, za primanjkljaj | 3-galaktozidazo in, po zadnjih podatkih, pomanjkanje encima a-sulfataza iduronosulfat [Swift Th, Mac Donald Th, 1976 .. ]. Bolezen poteka dlje časa v primerjavi z Hurler bolezni, simptomi niso tako izrazite, ni motnost roženice. V urinu, količina mukopolisaharidov povečal v krvnih limfocitov - granule ob metachromatic.
Mykopolisaharidoz tip III (Sanfilippo bolezen) - dedovanje avtosomno recesiven. V tkivih nakopičenih heparin sulfat. Obstaja pomanjkanje beta-galaktozidaza. Poleg tega je zmanjšana aktivnost v organih geparansulfatazy [Cain H. et al., 1976]. Nad somatskih sprememb prevladuje psihomotorične zastoj. Vendar pa glede na N. Cain et al. (1976), vsebnost GAG v jetrih povečala 12,5-krat po standardih, predvsem zaradi kopičenja heparan sulfata. Podobna snov najdemo v nevronih možganih, v stenah žil, vranici. Količina GAG urina povečala v krvnih limfocitov - metachromatic granul.
Mukopolisharidoze tipa IV (Morquio bolezen) -nasledovanie avtosomno recesiven. V tkivih kerataisulfat zamudo zaradi N-atsetilgeksozaminsulfatazy pomanjkljivost. Prevladujoči skeletnih sprememb in roženice zamegljenost. Zrnca limfocitov primanjkuje.
Tip Mykopolisaharidoz V (Sheye bolezen) -za trenutno predvideni kot varianta mukopolisaharidoze tipa I, ki se klinično kaže pri odraslih, saj encimske aktivnosti in L-iduronidazy nekoliko zmanjšan.
Tip mukopolisharidoze VI (Maroto bolezen - Lamy) - Dedovanje je avtosomno recesiven.
V tkivih zamuja dermatan. Obstaja pomanjkljivost encimskega 4-acetilgalaktozamin-4-sulfataze. Vse ostalo je podoben tip mukopolisharidoze bolezen I, vendar kršitve razvoja psihomotoričnih je zelo redko ali nikoli.
Tip mukopolisharidoze VII označena s pomanjkanjem encima beta-glukuronidaza. Tkivo nabira in se izloča v dermatan urina. Manifestacij bolezni so podobni mukopolisaharidozo tipa I, vendar ne tako izrazito.
mukopolisaharidoza običajno diagnosticira kliničnih simptomov, kot tudi na osnovi biokemičnih določanje GAG v urinu in prisotnosti metachromatic granul v krvnih limfocitov. Histokemična študija težko, saj GAG zelo hitro raztopijo v času posnetka tkiva in morfoloških sprememb maloharakterny. Če se pojavi bolezen v družini, je priporočljivo tudi amniocenteza, študijo plodovnice celic kulture in plodnika za prisotnost GAG.
Torej, na koncu, je treba poudariti, da so kljub prevladujejo kršitve izmenjav GAG v tako imenovanih "čistih" mukopolisaharidov motenj dejansko skupina bolezni, pri katerih je v večini primerov so kršitve mešanih tipa, mukopolisaharidov in gangliozidom.

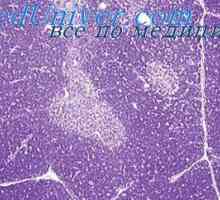 Kombinirana imunska z fermentopathy. nezelofa sindrom ali alymphocytosis
Kombinirana imunska z fermentopathy. nezelofa sindrom ali alymphocytosis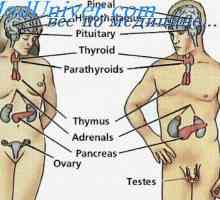 Hypophosphatasia plod. Orogenic displazija
Hypophosphatasia plod. Orogenic displazija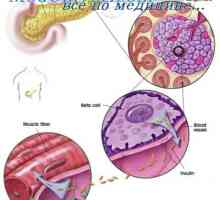 Avtosomno recesivno dedovanje. -X vezano dedovanje
Avtosomno recesivno dedovanje. -X vezano dedovanje Glyukotserebrozidoz Gaucherjeva bolezen. Glikosfingolipidoz Fabry bolezen
Glyukotserebrozidoz Gaucherjeva bolezen. Glikosfingolipidoz Fabry bolezen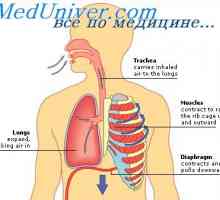 Mucolipidosis: mannozidoz in fucosidosis. Juvenilni sulfatidoz tip oustina
Mucolipidosis: mannozidoz in fucosidosis. Juvenilni sulfatidoz tip oustina Galaktoziltseramidoz Krabbe bolezen. Sulfatidoz metachromatic leykodistrosriya Scholz
Galaktoziltseramidoz Krabbe bolezen. Sulfatidoz metachromatic leykodistrosriya Scholz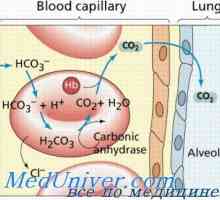 Amavroticheskaya prirojeno Idiotizam. Mucolipidosis
Amavroticheskaya prirojeno Idiotizam. Mucolipidosis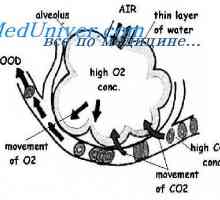 In leukodystrophies mukopolisaharidoze. Tveganje mukopolisaharidoze
In leukodystrophies mukopolisaharidoze. Tveganje mukopolisaharidoze Glycogenoses. Razvrščanje in manifestacije glikogenoza Gierke
Glycogenoses. Razvrščanje in manifestacije glikogenoza Gierke Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji
Glikogen ošpice bolezni shranjevanje, Andersen McArdl. Hersey bolezen, Thomson, kontejnerji Izvornih celic presajena za kopičenje bolezni in talasemije
Izvornih celic presajena za kopičenje bolezni in talasemije Vimizim odobren za zdravljenje Morquio sindroma
Vimizim odobren za zdravljenje Morquio sindroma- Gaucherjeva bolezen se nanaša na lipidov- akumulacije bolezni sfingolipidozam zaradi okvare gena,…
- Železo železov sulfat (Ferrosi sulfas). Besede:. Železov sulfat, železov sulfat, železo sulfuricum…
- Tardiferon (tardiferon) *. Depo Formulacija po železovega sulfata. Draže (retard), ki vsebuje 0,257…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
- Velika Medical Encyclopedia IC nevronet. bolezen
 Neyrometabolicheskie bolezen
Neyrometabolicheskie bolezen Mucolipidosis
Mucolipidosis Lizozomalnih bolezni pri otrocih
Lizozomalnih bolezni pri otrocih Mukopolisharidoze otroci, 1,2,3 vrsto zdravljenja, diagnozo, simptomi
Mukopolisharidoze otroci, 1,2,3 vrsto zdravljenja, diagnozo, simptomi