Anomalije ženskih spolnih organov. Sindromi Kaufman-mak-Cusick in Mayer-Rokitanskya-Kuster-Hauser
Prirojene anomalije genitalij najdemo v skoraj 3% živorojenih otrok. Te vključujejo anomalije jajcevodov, maternice, materničnega vratu in nožnice. Spekter nepravilnosti, vključno z agenesis, atrezija in izobraževalnih ovir. V pojmovanju, ženski zarodkov in diferenciacija primarnega Gonadni v ovary degeneracije Wolffian vodov in Müllerian diferencirajo v ženski reproduktivni sistem. Molekularno podlaga za razvoj ženskega reproduktivnega trakta človeka niso dobro razume.
Vemo veliko več o tem Razvoj v miših hvala za poskuse z "knockout" genov. Postopek Müllerian konjugata z apoptozo ali programirane celične smrti. Ta postopek je zelo urejeno, da pride vključuje več genov, vključno bcl-2 ščiti celice pred apoptozo. V odsotnosti izražanja gena ob pravem času, ko so bili združeni celice kanalizacija pripravljena resorpcijo lahko pride vztrajnost septum. Opisano človeški genetski sindromi, ki vplivajo na procese oblikovanja, diferenciacije in involucijo ženskega reproduktivnega progi.
K Genetsko sindromi oseba, vodi do nastanka patologije ženskega reproduktivnega trakta, so sindromi Kaufman-Mac Cusick in Mayer-Rokitanskya-Kuster-Hauser, kot tudi diabetesa pri mladih zrelega tipa, tip sindrom V. Kaufman-Mac Cusick - avtosomna recesivna bolezen, prvotno opisane v amiši verska skupnost značilna polydactyly, prirojeno srčno bolezen in gidrometrokolposom. Sindrom se lahko pojavi pri moških, vendar pa so se pokazale le anomalije strani in srčne bolezni.

nezmožnost preoblikovanja utero-vaginalni plošča kanal med embriogenezo vodi do tvorbe prečne predelne stene vaginalis ki povzroča maternične razteza in stiskanjem sosednjih organov. Te pregrade ponavadi lokalizirana v zgornji tretjini vagine. Takšno stanje se srečata s sindromom Bardet-BEADLE skupaj z retinopatije, invalidnosti, debelost, polydactyly, ledvic nepravilnosti in moški hipogonadizem učenje. MKKS gen kodira protein deluje kot šaperonina - predstavnik skupine proteinov, ki prispevajo k optimizaciji prostorske strukture drugih proteinov po izpustu iz ribosomov.
kako genske mutacije MKKS povzroči razvoj Kaufman sindrom-Mac Cusick ali bolj heterogeno sindrom, Bardet-Beadle, še vedno ni jasno.
Sindrom Mayer-Rokitanskya-Kuster-Hauserin je znan tudi kot sindrom Rokitanskya-Kuster-Hauser. Ta sindrom Müllerian aplazija, kjer je odkrita pomanjkanje telo maternice, materničnega vratu in zgornjih vagine odsekov. V večini primerov so bolniki - ženske z kariotip 46, XX, ki so shranjene na jajčnike in običajne zunanje spolovilo. povezana tudi s sindromom ledvične patologije in nepravilnosti včasih skeleta. Menijo, da je sindrom deduje avtosomno recesivno način, vendar še ni bila ugotovljena, odgovoren gen.
zdravljenje Gre za umetno ustvarjanje nove vagine z uporabo Dilators. MURCS simptom vključuje Müllerian aplazijo, ledvična agenesis, nenormalnosti vretenca, Klippel-Feilov sindrom in nizke rasti. To občasno sindrom neznano vedno vzročno genetsko anomalijo. Sindromi Fraser in Meckel-Gruber povezana tudi z Müllerian aplazije. Tip diabetes zrel mlada tipa V, - avtosomno dominantna patologija, Mutacija jeter jedrska faktor 1b (HNF-1b) so značilne nepravilnosti in ledvic aplazijo Müllerian. Mutacije tega gena lahko povzroči tudi Müllerian aplazijo in v odsotnosti mladoletniškega sladkorne bolezni.
Müllerian inhibitorni faktor, znan tudi kot AMG - predstavnik družine transformirajočega rastnega faktorja b (TGF-b), z gonad sintetiziranega. Po določitvi moške Gonadni v involucijo testisov transformacije Müllerian je eden od prvih simptomov spolne diferenciacije tipa moške. Trenutno dve vrsti opisal sindrom vztrajno Müllerian - I in II. Sem tipa I povzročajo mutacije AMG gen lociran na kromosomu 19. mutacija v tem genu zaznano pri 47% družinskih primerov persistentne Müllerian sindroma, se prenašajo v avtosomno recesivna način.
Tip II z mutacijami povzroča v genu Tip II receptorja AMH, ki izzove 38% trdovratno Müllerian sindroma. Vsebnost AMH seruma takšnih bolnikov v normalnih mejah. tip dediščine kot avtosomna recesivna. Bolniki so značilne kariotip 46, XY, in so običajno oblikovani zunanji moškega spolovila. Ti se lahko razvije kriptorhizem povezano z dimeljsko kriptorhizmom gryzhami- pogosto odkrijejo med operacijo.

 Fraser sindrom. Diagnoza in zdravljenje sindroma pri plodu Fraser
Fraser sindrom. Diagnoza in zdravljenje sindroma pri plodu Fraser Sistem moškega genitalnega kanal zarodka. Ženski reproduktivni sistem plod kanalizacija
Sistem moškega genitalnega kanal zarodka. Ženski reproduktivni sistem plod kanalizacija Spuščanje fetalnih jajčnike. Osnove genitalij zarodka
Spuščanje fetalnih jajčnike. Osnove genitalij zarodka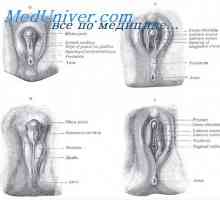 Anomalije maternice in vagine. testis plodu
Anomalije maternice in vagine. testis plodu Mullerian izločanja kanal. Razvoj moškega vzorca fetalni
Mullerian izločanja kanal. Razvoj moškega vzorca fetalni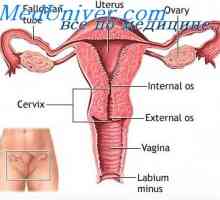 Fiziologija ženskih spolnih organov. Ženski hormonski sistem
Fiziologija ženskih spolnih organov. Ženski hormonski sistem Prirojene ledvic anomalije. ledvic agenesis
Prirojene ledvic anomalije. ledvic agenesis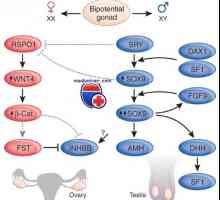 Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija
Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija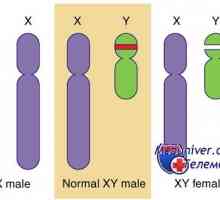 Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha
Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha Smrt, smrt jajčnih celic. povzroča apoptozo
Smrt, smrt jajčnih celic. povzroča apoptozo Cist idealno-genitalni sindrom. Genetika moške neplodnosti
Cist idealno-genitalni sindrom. Genetika moške neplodnosti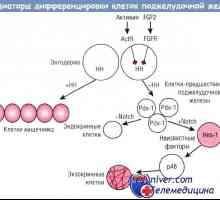 Uredba diferenciacije exocrine trebušne slinavke celic
Uredba diferenciacije exocrine trebušne slinavke celic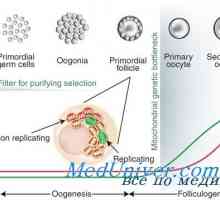 Zaposlovanje foliklov. Vpliv na folikulogeneze gonadotropinov
Zaposlovanje foliklov. Vpliv na folikulogeneze gonadotropinov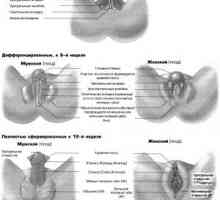 Razvoj spolnih organov pri plodu, ki ga tednih
Razvoj spolnih organov pri plodu, ki ga tednih Anomalije sečnega mehurja, sečnice in moški reproduktivni organi
Anomalije sečnega mehurja, sečnice in moški reproduktivni organi- Prirojene motnje spolnega differentsirovkizabolevaniya z kromosomskih nepravilnosti povzročajo.…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
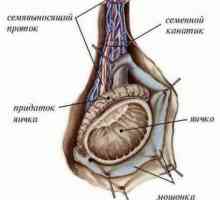 Razlikovanje med testisov in jajčnikov
Razlikovanje med testisov in jajčnikov Določitev obloge (vohun kot določitev faktorja testisov)
Določitev obloge (vohun kot določitev faktorja testisov) Müllerian anomalija
Müllerian anomalija Prirojene malformacije spolnih organov
Prirojene malformacije spolnih organov