Določitev obloge (vohun kot določitev faktorja testisov)
")
Študija moških s kariotipom 46, XX z zelo majhno Y-X-translokacije prepoznani gena, ki se nahaja proksimalno regijo psevdoautosomalnoy Y-kromosoma.
Ta gen je bil kloniran, osamljen in se imenuje SRY. Sry (analog gena humanega SRY pri miših) je izražen v mišjem spolnih tyazhah med 10,5 in 12,5 dni embrionalni razvoj, tik pred in med diferenciacijo testisov. Poleg tega, izbris ali mutacija gena človeškega sry odkrili pri 15-20% žensk z kariotip 46, XY, in XY-s popolno žlez dysgenesis. Najbolj očiten dokaz, da SRY - je dejavnik pri razvoju testisov, je dejstvo, da je miš Sry gen transfekciji v zarodkov z genotipom 46, XX vodi v nastanek testisov in moškega diferenciacije v teh živalih.
SRY gen kodira protein DNA vezavni ki vsebuje domeno sekvenco 80 amino podobni tistim, ki so bile ugotovljene v skupini proteina visoke mobilnosti (skupina visoko mobilnost - HMG). Ta domena se veže na DNK v nekem določenem zaporedju (A / CAAT-TAA). To upogne DNA in s tem omogoča interakcijo med proteini DNA vezavni, ki sproži transkripcijo osnovnega zaporedja gena ( "nižji stopnji" genov). Mehanizem delovanja SRY in njegovega nadaljnjega cilji niso znani, vendar SOX9 - nadaljnji genov kandidatke. Trenutno označene 4 osnovne celične funkcije sry beljakovin. Ti vključujejo indukcijo diferenciacije od Sertolijeva Celica, migracije primarnih ledvičnih celic genitalnega tubercle, tubercle proliferacije spolnih celic, oblikovanje žil v spolnih žlez iz moškega tipa, ki vključuje veliko število endotelnih celic iz mesonephros. Miši Sry spodbuja neposredno regulacijo Sox9 in vzpostavlja pozitivno povratno zanko, ki se aktivira in zatira Fgf9 Wnt4, da dosledno prispeva k oblikovanju testisov. V odsotnosti SRY genske ekspresije pri miših vodi do zmanjšanja Wnt4 Sox9 izražanja in Fgf9, ki vodi do tvorbe jajčnikov. Doslej ni opisana FGF9 mutacije pri ljudeh, zato je njegova vloga pri oblikovanju testisov pri ljudeh še vedno morali izvedeti. Večina mutacij opisane doslej pri ženskah s kariotip 46, XY gonadne dysgenesis da predstavljajo kršitve nukleotidnega zaporedja SRY gena, ki kodira DNA-vezavna domena (polje HMG) SRY protein. Vendar pa mutacije, ki vplivajo na DNK loki in transport molekul iz jedra, prav tako za posledico motnje v modih razvoju.
Ugotovljene drugih genov, ki so odgovorni za razlikovanje človeških mod. Heterozigotna mutiranje gena Wilmsov tumor (WT1), lokalizirana v območju 11p13, ne vodi le k razvoju Wilmsov tumor, temveč tudi prirojenih urogenitalnega abnormalnosti. Izklop WT1 gena pri miših vodi do apoptoze primarne ledvične blastema, kar vodi do pomanjkanja ledvic in spolne žleze. Tako WT1 - transkripcijski regulator, ki deluje na blastema primarni ledvic v zgodnjih fazah razvoja urogenitalnega trakta. Heterozigotna mutacije vodijo do človekovih sindromi Denys-Drash in Fraser, medtem najdaljši izbris gena in sosednji DNK vodi do nastanka Wilmsov tumor, aniridia, genitalnih motenj, duševne zaostalosti - WAGR sindroma.
Steroidogenske faktor-1 (SF-1) - jedrska receptorja povezano z regulacijo transkripcije mnogih genov, vključno s katalizatorji steroidogenske genov P450. V zadnjem času, v Krylov sod. objavljeni podatki, ki so lahko fosfolipidi med ligandi SF-1 in jedrskih receptorje tarčnih organov in urejajo njihovo dejavnost. SF-1 je proizveden v moških in ženskih spolnih tuberkla in v tkivih, ki so odgovorni za steroidogenezo, kjer je potrebna ta dejavnik za sintezo testosterona. Formulacija SF-1 se pojavlja v Sertolijeva Celica, kjer je faktor ureja antimyullerova hormona gen (AMG). SF-1 kodira zaporedje genoma sesalcev, genski homologna za Drosophila FTZ-F1. Je homozigotna izguba gena, ki kodira SF-1 pri miših vodi do apoptoze spolnosti kabel, ki povzroča nadledvične žleze ter žleze, zaradi motenih morfogenezo gonad in nadledvične žleze, kot so ženski in moški vrste. Tako je ta gen odločilno vlogo pri tvorbi organov-steroidnih izločajo (kot nadledvične žleze, moda in jajčniki). WT1 in SF-1 so aktivni v zgodnjih fazah razvoja spolne in Breg odločnosti kot jajčnikov in testisov. Heterozigotna in homozigotne mutacije SF-1 pri ljudeh vodi do tvorbe ženskega fenotip pri bolnikih z kariotip 46, XY, ki ga spremlja insuficience nadledvične žleze in žlez dysgenesis. Opisano heterozigotna nesmiselne mutacije SF-1 pri bolnikih z ženskim fenotip, genotip 46, XY, gonadne dysgenesis in normalno delovanje nadledvičnih žlez. Tako je za vse gene, ki sodelujejo pri spolnem razlikovanju, bistvenega pomena za tvorbo fenotipa je število ponovitev genov v celici (gena dozirne) v.
Gonadno dysgenesis pri bolnikih z genotipom XY ženski fenotip z razvojem lezij je bila ugotovljena pri ljudeh s podvojitvijo Xp21 odsotnosti SRY-funkcije, to procesno vsebuje DAX1 gen na kromosomu X. Mutacija ali izbris DAX1, ki kodira transkripcijski faktor, ki izhaja iz X-vezan prirojeno adrenalne hipoplazijo in hipogonadotropen hipogonadizem pri moških z genotipom 46, XY. Izbris ali mutacijo gena DAX1 ne vodi do kršitve diferenciacije mod pri moških. Podobno, podvojitev DAX1 gen ne zdi, da vplivajo na morfogenezo in delovanje jajčnikov pri ženskah, ki kariotip 46, XX. Miši z genotipom 46, XY (homologna miši), ali uporaba dvojne Dax1 delecijo gena vodi k spremembi spolne diferenciacije. To se zgodi z zmanjšanjem aktivnosti SRY, deloma kot posledica deaktivacije SOX9 v celičnih predhodnimi sestavinami sertoli. Tako je v mišje številko modela Dax1 igra ključno vlogo pri razvoju jajčnikov.
Gene DHH (dobeseden prevod "puščavski ježka") kodira signalne molekule, ki so na kromosomu 12q13.1 pri ljudeh. Homologni genski pri miših (DHH) začne se izrazi v celičnih predhodnimi sestavinami sertoli kmalu po SRY ekspresijo v Leydigovih celic. DHH je pomemben gen za razvoj in delovanje testisov pri sesalcih. V neki raziskavi je bilo ugotovljeno, da so imeli 3 od 6 bolnikov z kariotip 46, XY, gonadne dysgenesis in normalno ekspresijo SRY gena DHH homozigotne mutacije.
} {Modul direkt4
Kampomelicheskaya displazija je varianta displazije skeleta, povezanih z obračanjem na tla, ki je posledica žlez dysgenesis pri bolnikih z kariotip 46, XY. Gen, odgovoren za kampomelicheskuyu displazije (CMPD1), ki se nahaja na 17q24.3-q25.1. Mutacija enega alela SOX9 gen, povezan z sry (imenovali SOX, ker ima HMG polje, ki je več kot 60% homologna tisti sry gena), lahko pripelje do tako kampomelicheskoy displazija, in na XY-žlez dysgenesis in inverzije tla . Podvojitev SOX9 gen pri človeku in miših vodi prevrniti spolno kariotip pri posameznikih z XX. Možno je, da je SOX9 šele SR Y gena potreben za normalno moško diferenciacijo. V XY posameznikih z 9r- in 10q-delecije s popolno ali delno odsotnosti testisov, v Wolffian vodov in zunanjih genitalij odkrite. Haplo-odpoved DMRT1 gen - gen, povezan z "doublesex» Drosophila in "MAZ" ogorčice - lahko odgovoren za nepravilnosti v organogeneze testisov pri bolnikih s izbrisa 9P. Opisano več kot 22 bolnikov z kariotip 46, XY in monosomijo za 9P. Pri teh bolnikih obstaja vedno stigma 9P sindrom, kot nejasnih zhenskie- ali zunanjih spolovil, prisotnost Müllerian konstrukcij skupaj z nitastih disgenetichnymi spolne žleze ali modih. Zadnji podatki kažejo, da so lahko ženske z kariotip 46, XX in 9P sindrom delovanja jajčnikov normalno ali poškoduje. Pri teh bolnikih, pri 10q locus ni opredelila samo en gen, odgovoren za odpravo spola.
Ugotovljeno je bilo, da lahko pacienti s podvojitvijo 1r31-35 regijo, ki obsega WNT4 podvajanja genov, tvorimo s ženski fenotip kariotipom xy. Pričakovati je, da se razvoj testisov moten zaradi aktiviranja gena, ki ga podvojitev DAX1 Wnt4, kar blokira aktivacijo SF-1 in SOX9. V nasprotju s tem raziskovalna dejavnost Wnt4 (glodalski homolog) v transgenih miših so pokazali, da je prekomerno izražanje tega gena povezana s preprečevanjem sinteze testosterona in zmanjšuje dotok krvi v testisih, vendar ne vodi do spolnosti polov, kot je opisano pri ljudeh. Poleg tega je pri bolnikih z 46, XY prekomerno WNT4 klinično heterogeno fenotip: iz izoliranega kriptohidizem z ženskim zunanjih genitalij. In vitro študije so pokazale, da zmanjšanje Wnt4 steroidogenezo ga dominira SF-1 povzročenega prepisa. WNT4 izražen v jajčnikih in opredeljena kot pomemben signal določitev jajčnikov. Izražanje Wnt4 jajčnikov preprečuje migracijo steroid (androgen) -producing celic v notranjosti jajčnikov, zavira razvoj določenih modih krvnih žil (coelomic plovila), je zelo pomembna za razvoj kanala Mullerian in ohranjanje zarodnih celic. V mišjih samicah z ničelno mutacijo Wnt4 določi Müllerian hipoplazija in zarodnih celic, vendar je ugotovil razvoj Wolffian voda zaradi androgeni izločanje ovarijskih celic, ki proizvajajo steroidi. Wnt4 je potrebno tudi za normalen razvoj miške pochki- z ničelnimi mutacij Wnt4 umre kmalu po rojstvu z ledvično odpovedjo.
Literatura opisuje ženska z 46 genotip, XX in pomanjkanje struktur razvijanje Müllerian voda (netipično sindrom Mayer-Rokitanskya-Kuster-Hauser), ki je zmanjšano delovanje mutacije v WNT4 gena. Njena fenotip je podobno tistemu, opisanemu v miših s pozivom mutacije. Bolnik je imel enostransko okvaro agenesis in znake androgenizma, manifestira hude akne, ki potrebujejo protiandrogeno terapijo. Razvoj Wolffian vod ni bila ugotovljena. Vendar pa je preiskava velike skupine žensk s sindromom Mayer-Rokitanskya WNT4 ni pokazala mutacije.
Povečanje dokazi kažejo, da so mnogi geni odgovorni za razvoj yaichnikov- jajčniki ne tvorijo »privzeto« način. gen folistatin najbrž deluje podobno WNT4 v organogeneze jajčnikih. FoxL2 se domneva transkripcijski faktor, da imajo miši poporodna inhibira izražanje genov v testisih, ki je odgovoren za razlikovanje moškega tipa. FoxL2 mutacije privede do preoblikovanja v dvajsetem nadstropju v "hornless kozo" in FOXL2 mutacij prizadetih žensk z kariotip 46, XX vodi do motenj delovanja jajčnikov, z ali brez blepharophimosis njega.
 Nepravilnosti SOx genov in TVH Holt-Oram sindroma. Dejavniki fibroblastov rasti
Nepravilnosti SOx genov in TVH Holt-Oram sindroma. Dejavniki fibroblastov rasti Spermatogeneza. Zorenje semenčic v testisih
Spermatogeneza. Zorenje semenčic v testisih Anomalije ženskih spolnih organov. Sindromi Kaufman-mak-Cusick in Mayer-Rokitanskya-Kuster-Hauser
Anomalije ženskih spolnih organov. Sindromi Kaufman-mak-Cusick in Mayer-Rokitanskya-Kuster-Hauser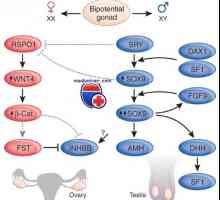 Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija
Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija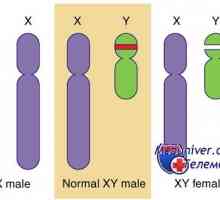 Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha
Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha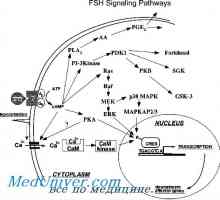 Gonadotropin receptorje. Struktura in funkcija
Gonadotropin receptorje. Struktura in funkcija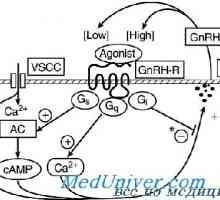 Receptor za gonadoliberin. Mutacije GnRH receptorja
Receptor za gonadoliberin. Mutacije GnRH receptorja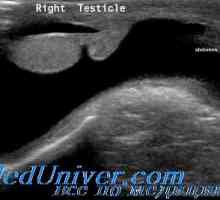 Spermatocele in hydrocele pri otrocih. testisov tumorjev
Spermatocele in hydrocele pri otrocih. testisov tumorjev Znanstveniki so ugotovili vzrok moške agresije
Znanstveniki so ugotovili vzrok moške agresije- BDNF debelost in genetika
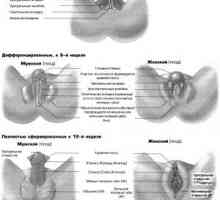 Razvoj spolnih organov pri plodu, ki ga tednih
Razvoj spolnih organov pri plodu, ki ga tednih- Prirojene motnje spolnega differentsirovkizabolevaniya z kromosomskih nepravilnosti povzročajo.…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
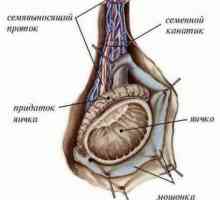 Razlikovanje med testisov in jajčnikov
Razlikovanje med testisov in jajčnikov Genetska sex, določitev
Genetska sex, določitev Res hermafroditizem
Res hermafroditizem Primarni hipogonadizem pri moških in ženskah: Zdravljenje
Primarni hipogonadizem pri moških in ženskah: Zdravljenje Dvostranska anorchidism (izginile moda sindrom)
Dvostranska anorchidism (izginile moda sindrom) Fiziologija moškega reproduktivnega sistema
Fiziologija moškega reproduktivnega sistema Miotonična distrofija
Miotonična distrofija Xy kariotipa 46 moških
Xy kariotipa 46 moških