Xy kariotipa 46 moških

Vsebina
- Pure gonadno dysgenesis (suayra sindrom)
- Vztrajanje müllerian
- Anorchidism
- Kršitve sintezo in delovanje androgenov
- Kršitve sintezo in delovanja lh
- Kršitve sinteze androgenskega
- Prirojena nadledvične hiperplazija lipondnaya
- 3-hidroksisteroid-dehidrogenaze pomanjkljivost
- Hydroxylase pomanjkljivost 17h
- $ 17-hidroksisteroid-dehidrogenaze pomanjkljivost
- Pomanjkanje 5a-reduktaze
- Napake androgenskega receptorja
- Popolna odpornost androgen (testisov feminizacija)
- Delna odpornost na androgenov (delna androgenskega sindrom neobčutljivost)
- Ne endokrinih vzrokov razvojne motnje moških spolnih organov
Kariotip 46, XY.
Pure gonadno dysgenesis (Suayra sindrom)
Obstajata dve teorije patogenezo te bolezni. Običajno se zarodnih celic začnejo vplivati na razvoj spolnih žlez šele po selitvi v spolnih kabli. Menijo, da pri nekaterih bolnikih s čistim žlez dysgenesis to gibanje ne zgodi, in namesto mod so oblikovali tyazhevidnye spolnih žlez. Ostali bolniki kažejo mutacije genov, ki so odgovorni za razvoj testisov, ki izhajajo iz zarodnih celic ne prodrejo v snov možganov brezbrižni spolne žleze ostanejo v kortikalnih in hitro umre. V navzočnosti Y kromosomov reproduktivnih celic popolnoma ubili. Pri 15% bolnikov s čistimi žlez dysgenesis našel SRY genskih mutacij, ostalo, očitno je, da obstajajo tudi druge mutacije genov, ki sodelujejo v modih razvoj. Opisano-X mrežnimi oblika bolezni je očitno posledica dejstva, da so geni, ki se nahajajo pod SRY kritično razlikovanje gonad. Pri teh bolnikih obstaja normalen razvoj Müllerian in oblikovani ženski fenotip.
Pri bolnikih s čisto žlez dysgenesis 46, XY ženski fenotip značilnost in zamude spolnega razvoja. V nasprotju s tem pa pri bolnikih s Turnerjevim sindromom, imajo dokaj visoko rast, saj, prvič, ni cone rasti zdravljenje epifiznih za dolgo časa ostala odprta zaradi pomanjkanja spolnih hormonov, in drugič, v Y-kromosoma so geni, ki pospešujejo rast . Ravni gonadotropin sproščujočega hormona v krvi znatno povečala. V laboratorijski študiji kažejo kariotip 46, XY. Tveganje maligne transformacije tyazhevidnyh žlez ženskah z boleznijo znaša 25 do 35% - višja kot pri drugih oblikah žlez dysgenesis z Y-kromosoma. Po diagnozi je potrebno odstraniti spolnih žlez. Včasih zarodnih celic tumorjev v puberteti začnejo izločati testosteron ali estrogen, ki vodi do različnih stopenj feminizaciji ali virilization.
Vztrajanje Müllerian
Pri normalnem migracije spolno diferenciacijo moškega tipa in diferenciacijo zarodnih celic mod začne takoj po aktivaciji in od njih-gena pod nadzorom genov. Kasneje začne sintetizirano Anti-Müllerian hormon, je lokalno zavira razvoj njihovih derivatov. Müllerian obstojnost je lahko posledica mutacije gena antimyullerova hormona (kot posledica ravni faktorja v krvi je zelo nizka ali pa ne zazna) ali okvare receptorja proteina. Kot rezultat, bolniki z moško fenotip tudi maternice in jajcevodih. V odsotnosti drugih genskih motenj testisov razvija normalno in dobimo zadostno količino androgenov.
Pri bolnikih z dolgotrajno Müllerian zaradi kršitve postopka testisov rodu običajno pojavi enkrat ali dvostransko kriptorhizem in dimeljske kile. Včasih kažejo malformacij semenovod. Odstranitev maternice in vagine je povezan z velikim tveganjem poškodb semenovod in kasnejše neplodnosti.
anorchidism
Ko anorchidism testisi začeti normalno razvija in proizvaja anti-Müllerian hormon, vendar pred sintezo testosterona opraviti atrofijo. Prej je bilo prepričanje, da so razlog za to so žilne bolezni ali spremembe v okoliško tkivo, vendar pa glede na dvostranski lezije, se zdi bolj verjetno motnje na molekularnem nivoju. Po regresijsko Müllerian testosterona sintezo in diferenciacija volfovyh kanal ne pride.
Fenotip anorchidism so različni in odvisni od stopnje spolnega razvoja, ki se je zgodil atrofijo mod. Praviloma so bolniki rodil z žensko fenotip, in starši jih prinese k zdravniku, ker ne pride v puberteto. V laboratorijski raziskavi razkrila kariotip 46, XY in visoko raven gonadotropini hormonov. Ko revizija medenica ne zazna žlez zvoka ali derivati genitalne kanale.
Kršitve sintezo in delovanje androgenov
Pri bolnikih z kariotip 46, XY in krši delovanja androgenov differeniirovka testise običajno poteka normalno in antimyullerova izločanje hormona ne trpi. Vzroki za sintezo in delovanja androgenov:
- Bolezni sinteze in ukrepi LH;
- Pomanjkljivost encimov, ki sodelujejo pri sintezi androgeni;
- pomanjkljivosti v androgene receptorje.
Kršitve sintezo in delovanja LH
Leydigove celice se nahajajo na Ai receptorski vezavni teh receptorjev na ligand sproži sintezo androgenov v modih. stimulacije receptorjev LH zaradi visokih ravni hCG v serumu pri materi poteka pred pričetkom izločanja LH, ki ga hipofize zarodka. Bolniki z 46.XY kariotip Zaznali receptorjev genskih mutacij GnRH in LH podenoto gena. Med nosečnostjo se raven hCG je dovolj visoka, da delujejo sintezo testosterona v modih. Ob rojstvu otroka je normalen moški fenotip, tako da je diagnoza je samo v adolescenci, ko iščejo zdravniško pomoč zaradi zamude pubertete. LH gena receptorja mutacije pri bolnikih z kariotip 46, XY verjetno motijo tvorbo moškega fenotip, saj hCG ne veže na LH receptorje in ni sposoben stimulirati sintezo androgen pri plodu. Simptomi bolezni so podobni odpornost na androgenov.
Kršitve sinteze androgenskega
Ko odpoved ali odsotnost encimov, ki sodelujejo pri sintezi androgene je androgena izločanje zmanjša in razvoj genitalnih vodov in zunanje genitalije moten. Sinteza androgenov, se lahko razdelijo v različnih fazah, in stopnja okvare določenega encima je lahko tudi drugačna - od manjšega zmanjšanja aktivnosti do njene popolne odsotnosti. Klinična slika je raznolika: obstajajo enostranski ali, pogosteje, dvostranski kriptorhizem, mikropena. Vse bolezni so podedovali autosomnoretsessivno.
Prirojena nadledvične hiperplazija lipondnaya
Ta redka bolezen, ki jo povzroča genetsko določene motnje steroidogenezo v zgodnji fazi. Akutna steroidogenske odziv urejeno holesterola za od zunanje do notranje mitohondrijske membrane in sprožilca tega procesa je protein zvezda. Stalna steroidogenske zmogljivost CYP11A1 gen prepis je določena. Star genske mutacije krši ni vse steroidogenez- majhen delež Star neodvisne steroidogenezo izgubljeno kasneje v zvezi s sekundarno poškodbe celic, ki pojasnjuje možnost pozno, le nekaj mesecev po rojstvu, znaki insuficience nadledvične žleze. Podoben sindrom je redka povzročajo mutacije CYP11a (opisan dve varianti) kodira P450ssc.
3-hidroksisteroid-dehidrogenaze pomanjkljivost
3 -gidroksisteroidtsegidrogenazy pomanjkanje vodi do povečane ravni dehydroepiandrosterone in nižjih ravni spolnih hormonov - testosterona in androstenediona. Za bolezen označena z izgubo sindrom sol, hiponatriemija in hiperkaliemija znakov. Če čas ne bi diagnozo in začeti zdravljenje, lahko novorojenčka razvije insuficience nadledvične žleze. -gidroksisteroidtse encima 3-hidrogenazo je kodiran z dvema genov - HSD3B1 in HSD3B2. Pri bolnikih s pomanjkanjem encima razkrivajo mutacij HSD3B2 gen.
Hydroxylase pomanjkljivost 17h
sindrom pomanjkljivost 17a-hidroksilaze - redka genetska motnja steroidni biosintezi, kar ima za posledico zmanjšanje proizvodnje kortikosteroida in sintezo spolnih steroidnih hkrati povečuje mineralokortikoidov prekurzorje. Citokromom P450c17 šteje bifunkcionalno encim, ki imajo tako 17 in -gidroksilaznoy 17,20-liaze in kodiran s CYP17 gena. Pomanjkanje različnih encimskih funkcij zaradi različnih mutacij v genu. P450c17 zazna v žarka in mrežasto območja v nadpochechnikov- lupina zona glomerulozni ustreza steroidni biosintezi brez odcepov, tako mineralokortikoidov sinteze ni moteno. Neto Steroidogeneza cona, nasprotno, je razdeljena v zgodnji fazi, in proizvodnja androgen ne poteka, zaradi česar, zunanja spolovila, ne glede genetskega spola so ženske. Lahko razvije hipokaliemijo, in hipertenzijo zaradi kopičenja prekurzor steroidnih hormonov in tvorjenje mineralokortikoidov presežek (npr deoksikortikosteron). Ravni ACTH, FSH in LH v krvni plazmi teh bolnikov je običajno povišana. Pri bolnikih z kariotip 46, XY maternice in jajčnikov ne obarvajo. Zdravljenje z glukokortikoidi in diuretiki s kalijem pomaga normalizirati krvni tlak in preprečuje izgubo kalija.
$ 17-hidroksisteroid-dehidrogenaze pomanjkljivost
17. odpoved-hidroksisteroid-idrogenazy denarja vodi do motenj preoblikovanja androstenediona v testosterona je označena s kopičenjem dehidroepiandrosterona in androstenediona ter zmanjša sintezo testosterona, in, končno, dihidrotestosteron. Brez zdravljenja se presežek androstenediona pretvorimo estrona, ki spodbuja razvoj mlečnih žlez. Ugotovili smo, večletne mutacije -gidroksisteroiddegidro genov 17-dehidrogenaze.
Pomanjkanje 5a-reduktaze
Pri normalnem spolne diferenciacije Leydigove celice proizvajajo testosteron, ki v tarčnih celicah z učinkovanjem 5-reduktaze pretvori v močnejši androgen, dihidrotestosteron. Testosteron in delujejo dihidrotestosteron na iste receptorje androgenov dihidrotestosteron, a manjši disociacijsko konstanto, pri čemer veže močnejši na receptor in ima močnejši učinek. Na 5 stopenj reduktaze pomanjkanje testosterona normalne ali zvišane. Bolezen povzroča doslednih kršitve zunanjih genitalij. Testosteron zagotavlja normalen razvoj volfovyh kanale, saj je 5-reduktaze niso vključene v ta proces.
Pred puberteto razlikovati odpoved 5a-reduktaze iz sindrom androgene odpornosti zelo težko, še posebej, če so jajca odstranijo pred nastopom pubertete. Ko shranite moda v najstniških letih pogosto pojavlja bolnikov Virilization. Tako kot v normalnem puberteti, v tem obdobju pa je močno povečanje testisov sinteze testosterona. Kot rezultat, kljub stalni pomanjkanju 5a-reduktaze, količina testosterona zadošča za zagotavljanje lokalne učinek na ciljne celice. Zunanje genitalije začeli preoblikovati moški tipu- mogoče oblikovati polnopravni penis. Ta pojav je opaziti v velikem družinskem študije v Dominikanski republiki, kjer so bili ti bolniki imenovano guevedoces, kar pomeni, moda v 12 letih. Shranjevanje ženski fenotip pri odraslih s pomanjkanjem 5a-reduktaze zgodi zelo pogosto.
Encim 5 reduktaze je kodiran z dvema genov - srd5a1 in SRD5A2. SRD5A2 gen izražen v genitalnem področju, zato je bila njegova mutacije privede do klinični sliki zgoraj opisano. SRD5A1 gen izražen v kožo, predvsem lasišče.
Napake androgenskega receptorja
Posledice odpornosti na androgene pojavijo v fazi zunanje genitalije. Pri teh bolnikih, Müllerian atrophied testisi razvija normalno in izločajo zadostne količine testosterona, ki je v perifernih tkivih pretvori v dihidrotestosteron. Vendar androgen receptorje so odsotni ali ne more, da se veže na androgene-mi, tako da normalen moški fenotip ne razvijajo.
Oblika in androgen gena receptorja mutacije proučili precej dobro. To se nahaja na daljšem kraku kromosoma X in vsebuje 8 eksonov v vsaki od katerih se lahko mutacije (predvsem točka) in vodi do različnih oblik sindroma androgenskega odpornostjo - blaga za dokončanje. Oba popolno in delno odpornost na androgena dedna recesiven, sklopka z X-kromosoma. V družinah različnih bolnikov, kažejo edinstvene mutatsii- iste mutacije zazna zelo redki, zato razviti metodo za ugotavljanje genetsko odpornost na androgenov zaradi napake v en sam gen ne more biti. Različne mutacije privede do neenakomerne receptorje napakah. Najpogosteje je kršil nukleotidno zaporedje kodira gormonsvyazyvayuschy domeno. Druge mutacije vplivajo na domeno DNA veže. V tem primeru je funkcija androgene receptorje ne trpi, ampak kompleks hormon - receptorja ne more komunicirati z določenih spletnih mestih DNA (postreceptor hibe). pomanjkljivosti Intenzivnost receptorja razlikuje od popolnega androgen neobčutljivost na skoraj normalno hormonsko prenos signala.
Popolna odpornost androgen (testisov feminizacija)
Običajno razvita moda razporejene v trebušni votlini, dimeljske kanal ali sramnih ustnic, na spermatogeneze pa odsoten ali je nepopolna. Po koncu spolnega zorenja jajčec je treba odstraniti, ker je tveganje njihovega maligne transformacije je 2-22%. Včasih se oteklina pojavi pred nastopom pubertete, pa se je zgodaj odstranitev mod proizvajajo ni priporočljivo, saj so pomembni za razvoj ženskih sekundarnih spolnih značilnosti in psihosocialne nadaljnje prilagajanje. Blokiranje delovanje androgenov brez povezave, lahko to mlečna žleza doseže velik razmerov- kjer so sestavljene predvsem iz maščobnega tkiva.
Delna odpornost na androgenov (delna androgenskega sindrom neobčutljivost)
To je redka bolezen, kot na modih feminizaciji. Klinični znaki sindroma spreminjala. Bolniki so običajno postavljeno kot dekleta. V najstniških letih je sramne rast las poveča in poveča klitoris.
Fenotip bolnikov lahko moški ali ženske, s masculinization izraženo v različni meri. Nekateri bolniki našel hypospadias, drugi pa ne razlikuje od zdravih moških in trpi samo ginekomastija in neplodnost.
Ne endokrinih vzrokov razvojne motnje moških spolnih organov
Med normalno spolno diferenciacijo moškega vzorca je mogoče opaziti tudi motnje spolnega razvoja. Običajno je izoliran pomanjkljivosti: pomanjkanje mod ali penisa, penis displazija (podvojitev epispadias, kongenitalna kloake), nenormalno položaj glede na modih penisa. Možni vzroki: okoljski dejavniki, plodovnica zoženje, kromosomske nepravilnosti, genetske napake v maternici.
 Kaj določa dednost? Primarne zarodnih celic
Kaj določa dednost? Primarne zarodnih celic Jajc v lupini. zarodnih celic
Jajc v lupini. zarodnih celic Bookmark spolne žleze. Razvoj zarodnih celic
Bookmark spolne žleze. Razvoj zarodnih celic Bookmark zarodnih celic. določitev kromosomskih sex
Bookmark zarodnih celic. določitev kromosomskih sex Razvoj notranjih spolnih organov pri plodu. Zaznamek spolnih žlez plod
Razvoj notranjih spolnih organov pri plodu. Zaznamek spolnih žlez plod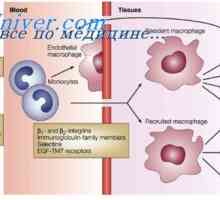 Mullerian izločanja kanal. Razvoj moškega vzorca fetalni
Mullerian izločanja kanal. Razvoj moškega vzorca fetalni Diagnoza in zdravljenje Turnerjev sindrom. Pure gonadno dysgenesis
Diagnoza in zdravljenje Turnerjev sindrom. Pure gonadno dysgenesis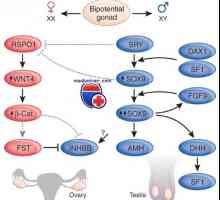 Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija
Mutacija in gen podvajanje dax1, sox9. spol neskladje xy genotip in kampomelicheskaya displazija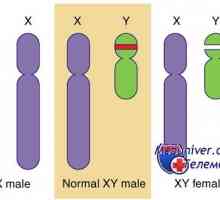 Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha
Genetske motnje spolnih žlez. Geni sry, WT1 in sindromi Fraser in Denis-dresha Cist idealno-genitalni sindrom. Genetika moške neplodnosti
Cist idealno-genitalni sindrom. Genetika moške neplodnosti Dve presenetljivo celice: jajce in sperma
Dve presenetljivo celice: jajce in sperma Razvoj spolnih organov pri plodu, ki ga tednih
Razvoj spolnih organov pri plodu, ki ga tednih- Prirojene motnje spolnega differentsirovkizabolevaniya z kromosomskih nepravilnosti povzročajo.…
- Zdravje Enciklopedija, bolezni, zdravila, zdravnik, lekarna, okužba, povzetki, spol, ginekologije,…
 Res hermafroditizem
Res hermafroditizem Hypergonadotrophic hipogonadizem
Hypergonadotrophic hipogonadizem Primarni hipogonadizem pri moških in ženskah: Zdravljenje
Primarni hipogonadizem pri moških in ženskah: Zdravljenje Določitev obloge (vohun kot določitev faktorja testisov)
Določitev obloge (vohun kot določitev faktorja testisov) Kariotipa 46 xx ženska
Kariotipa 46 xx ženska Amenoreja jajčnikov
Amenoreja jajčnikov Prirojene malformacije spolnih organov
Prirojene malformacije spolnih organov