Prirojeno in dedno tubulopatiji pri otrocih
Video: Vzroki za avtizem: genetika, gen za avtizem, cepivo DTP, gluten
Vsebina

Tubulopatije - bolezen s primarnimi lezij vodilni cevast, v kateri pretrgana membrana prevoz različnih snovi v ledvičnih cevkah.
Video: Instincts, otroci, stres, izobraževanje - Jacque Fresco - Venus Project
Pri novorojenčkih zaznanih klinično izoliranega tubulopatiji, ki so večinoma spremljajo poliurija in motnje elektrolitov koncentracij v plazmi. Ti vključujejo Bartterjevega sindrom, Liddle, Lowe, diabetes insipidus, primarni gipoaldosteronizm tipa I, hiperkalciemijo o "zgodnjem otroštvu" ali cevastega tipa acidoza IV. Razen Bartterjevega sindroma in Lowe, morfološke spremembe v ledvicah na teh tubulopatije manjkajočega ali označena z razvojem nefrokalcinozi.
Video: diagnoza prirojeno in dedno patologije seminarju
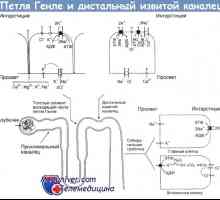
Bartterjevega sindroma - tubulopatiji, hipokalemija kaže, chloropenia, metabolična alkaloza in hiper-reninemiey z normalnim krvnim tlakom. Obstajajo tri oblike sindroma: novorojenčka, klasika in Gitelman sindromom (pri novorojenčkih niso našli). Novorojenčkov in klasične Bartterjevega sindromi včasih dedna avtosomno recesivno, vendar v večini primerov le občasno. Označene z dvema genotip podtip novorojenih oblike sindroma. Podvrsta sem posledica mutacije v genu kotransportnom natrijevega in kalijevega klorida (SLC12A1 lokus na daljšem kraku kromosoma 15-15q15-21), podtip II - ROMK mutacij v genu (KCNJ1 locus na daljšem kraku kromosoma 11 (11q24-25) za klasično Bartterjevega sindroma. označen s mutacije v genu natrijevega kanala (locus CLCNKB na kratkem kraku kromosoma 1-1r36). v obeh oblikah je polyhydramnios matere in dojenčkih pogosto rodil prezgodaj, z rojstvom je lahko ogromen poliurija. v študiji amnijske tekočine v rojstvom obdobju je zaznan SW lichenie vsebnost kloridov, kot je ultrazvočnimi nefrokalcinozi, hidronefroz in včasih kot posledica kroničnih sečevod poliurija določena.
Novorojenčka oblika je diagnosticirana kmalu po rojstvu, klasična - pri novorojenčkih in dojenčkih, starih do dveh let.
Video: Prirojena bolezen otrok - je izziv za starše?
Mikroskopsko opazili, skupaj z hiperplastični nefrokalcinozi jukstaglomerulni aparat (pathognomonic znak) redkeje - hiperplaziji, medularni intersticijskih celic, včasih - hyalinosis glomerulne vacuolization apical epitelnih cevkami, cevasto atrofijo in intersticijsko fibrozo. Smrtnost in umrljivost opazili v neobdelanem Bartterjevega sindroma. Pri zdravljenju otroka stanju bistveno izboljšalo, vendar je dolgoročna prognoza ostaja negotova, saj se postopoma napreduje počasi CRF.
Lowe sindrom (oculocerebrorenal sindrom) - redka genetska bolezen je-X vezana recesivna način dediščine. Sindrom je posledica mutacije OCRL1 gena na kromosomu X (Xq25-26). Ženske - nosilci za neobičajno gena imajo sive mrene, vendar imajo normalno ledvic in nevrološke funkcije. Bolezen je označen s kombinacijo prirojene sive mrene ali glavkom, huda duševna zaostalost, hipotonija z ledvično tubularno disfunkcijo.
Intenzivnost in čas cevastega disfunkcije so spremenljivi. Delovanje ledvic in struktura ob rojstvu lahko normalno. Proksimalni Cjevčica motnje pogosto začne v starosti od 3-12 mesecev. Mikroskopska struktura ledvic v plodovih in novorojenčkih običajno normalno. Vendar pa že so v prvih mesecih življenja opaziti širitev cevkah, ki jih atrofije in beljakovin valji v lumen. Glomerula običajnih novorojenčkov in potem ustvarjanje skleroze (fokalna ali globalna glomeruloskleroze), GBM zgosti. Elektronska mikroskopija navedli združitev noge podocytes in cevasto mitohondrijske škodo, vendar pa ni jasno, ali so primarno v zvezi s cevnim disfunkcijo.
 Galaktozemije
Galaktozemije Nega v sili v hipokaliemijo
Nega v sili v hipokaliemijo- Avtizem se naučijo prepoznati otroke na šestih mesecev
 Diagnoza, diferenciacija, prognozo in zdravljenje diabetes insipidus
Diagnoza, diferenciacija, prognozo in zdravljenje diabetes insipidus Diabetes insipidus: vzroki, klinična
Diabetes insipidus: vzroki, klinična Sekundarni aldosteronizem. Gormonalnoaktivnye tumor nadledvične skorje
Sekundarni aldosteronizem. Gormonalnoaktivnye tumor nadledvične skorje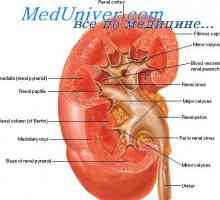 Tvorba urina v ledvicah. GFR
Tvorba urina v ledvicah. GFR Cistinoze otroci. Lo in Fanconijeve sindromi
Cistinoze otroci. Lo in Fanconijeve sindromi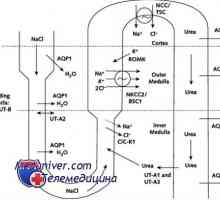 Diagnoza ledvično diabetes insipidus pri otrocih. zdravljenje
Diagnoza ledvično diabetes insipidus pri otrocih. zdravljenje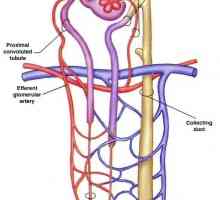 Distalnokanaltsevy acidoza pri otrocih. klinika
Distalnokanaltsevy acidoza pri otrocih. klinika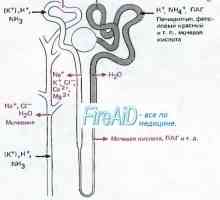 Acidoza ledvičnih tubulov. Proksimalnokanaltsevy
Acidoza ledvičnih tubulov. Proksimalnokanaltsevy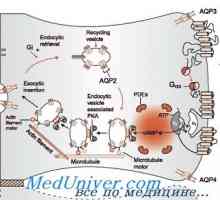 Rahitisa s cevastim acidoze. Nefrogeni diabetes insipidus pri otrocih
Rahitisa s cevastim acidoze. Nefrogeni diabetes insipidus pri otrocih- Gitelman sindrom pri otrocih. Diagnoza in zdravljenje
 Sindrom barter pri otrocih. Diagnoza in zdravljenje
Sindrom barter pri otrocih. Diagnoza in zdravljenje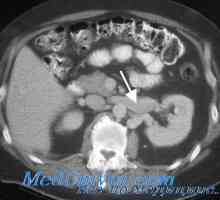 Tromboza ledvičnih žil pri otrocih. Diagnoza in zdravljenje
Tromboza ledvičnih žil pri otrocih. Diagnoza in zdravljenje Sekundarni in prirojene nefrotski sindrom pri otrocih. diagnostika
Sekundarni in prirojene nefrotski sindrom pri otrocih. diagnostika- Zdravljenje bolezni ledvic,
 Pseudohyperaldosteronism
Pseudohyperaldosteronism Tubulnega acidoza: simptomi, vzroki, zdravljenje, simptomi
Tubulnega acidoza: simptomi, vzroki, zdravljenje, simptomi Fanconijev sindrom: Simptomi, Zdravljenje
Fanconijev sindrom: Simptomi, Zdravljenje Drugo patologija pri novorojenčkih
Drugo patologija pri novorojenčkih